گفتگوها Clin Neurosci. 2013 Dec;15(4):431-43.
چکیده
علیرغم اهمیت عوامل روانشناختی متعدد، اساسا اعتیاد به مواد مخدر شامل یک فرایند بیولوژیکی می شود: توانایی قرار گرفتن مکرر در معرض مواد مخدر از سوء استفاده برای ایجاد تغییر در یک مغز آسیب پذیر که باعث جستجو و مصرف اجباری و مصرف دارو و از دست دادن کنترل می شود بیش از مواد مخدر استفاده می کنند که یک حالت اعتیاد را تعریف می کنند. در اینجا، ما انواع سازگاری های مولکولی و سلولی را که در ناحیه مغز خاصی رخ می دهد به منظور کاهش اختلالات رفتاری مرتبط با اعتیاد بررسی می کنیم. این شامل تغییرات در بیان ژن به دست می آید به وسیله مکانیزم های اپیزیونیک، پلاستیک در عملکرد نوروفیزیولوژیک نورون ها و سیناپس ها، و پلاستیک های مربوط به مورفولوژی نورون ها و سیناپسی که به طور ناگهانی بوسیله سیگنالینگ عامل نوروپاتیک تغییر یافته است. هر یک از این نوع تغییرات ناشی از مواد مخدر را می توان به شکل "حافظه سلولی یا مولکولی" مشاهده کرد. علاوه بر این، قابل توجه است که اکثر اشکال انعطاف پذیری مربوط به وابستگی بسیار شبیه به نوع پلاستیسیته است که با اشکال کلاسیک بیشتری از "حافظه رفتاری" مرتبط شده اند، شاید این نشان دهنده محدودیت مجدد سازوکارهای سازگار موجود در نورون ها در مواجهه با محیط زیست چالش ها. در نهایت، سازگاری های مولکولی و سلولی مربوط به وابستگی به اکثر مناطق مشابه مغز است که بین اشکال کلاسیک بیشتری از حافظه به میان می آیند، که مطابق با دیدگاه هایی است که خاطرات غیر طبیعی رؤسای مهم سندرم های اعتیاد هستند. هدف این مطالعات که هدف اصلاح مولکولی و سلولی مبتنی بر اعتیاد به مواد مخدر است، در نهایت، آزمایش های تشخیصی مبتنی بر زیست شناختی و همچنین درمان های موثر تر برای اختلالات اعتیاد را توسعه می دهد.
معرفی
اعتیاد به مواد مخدر، که می تواند به عنوان جستجوی و مصرف مواد مخدر با وجود عواقب وحشتناک یا از دست دادن کنترل مصرف مواد مخدر تعریف شود، ناشی از تغییرات ناشی از داروهای طولانی مدت است که در برخی مناطق مغز رخ می دهد.1 با این حال، بعضی از افراد در معرض ابتلا به داروهای مکرر دارو قرار می گیرند، در حالی که دیگران قادر به استفاده از مواد مخدر هستند و از سندرم اعتیاد فرار می کنند. عوامل ژنتیکی تقریبا٪ 50٪ از این تغییرات فردی در آسیب پذیری اعتیاد را تشکیل می دهند و این میزان وراثت پذیری برای همه طبقات عمده داروهای اعتیاد آور، از جمله محرک ها، مواد مخدر، الکل، نیکوتین و کانابینوئید ها صادق است.2 هنوز مشخص نشده است که بسیاری از ژن هایی که این خطر ژنتیکی را تشکیل می دهند، احتمالا به سبب دخالت احتمالا صدها تنوع ژنتیکی که در یک فرد تنها در معرض آسیب پذیری اعتیاد قرار می گیرند (یا در سایر افراد، مقاومت) است.
50٪ خطر دیگر اعتیاد به دلیل انبوهی از عوامل محیطی است که در طول زندگی رخ می دهد و در اثر تعامل با ترکیب ژنتیکی فرد ، وی را در برابر اعتیاد بیشتر یا کمتر آسیب پذیر می کند. انواع مختلفی از عوامل محیطی در اعتیاد نقش دارد ، از جمله استرس های روانی - اجتماعی ، اما تا حد زیادی قوی ترین عامل قرار گرفتن در معرض داروی سو of مصرف است. نشان داده شده است که برخی از داروهای "دروازه ای" ، به ویژه نیکوتین ، آسیب پذیری فرد را نسبت به اعتیاد به داروی دیگر افزایش می دهد.3 افزون بر این، شواهد افزایشی وجود دارد که علیرغم طیف وسیعی از خطرات ژنتیکی برای اعتیاد در سراسر جمعیت، قرار گرفتن در معرض دوزهای کافی از دارو برای مدت زمان طولانی می تواند فردی را که دارای بارگذاری ژنتیکی نسبتا پایین است به یک معتاد تبدیل کند.4
پیشرفت های بزرگی در طی دو دهه گذشته در شناسایی هر دو منطقه گسسته مغز، مهم در میانجیگری سندرم اعتیاد، و همچنین انواع تغییرات در سطوح مولکولی و سلولی است که مواد مخدر در این مناطق به آنها منجر می شود تا جنبه های کلیدی را زیر پایه گذارند از اعتیاد1,5 مدار که بیشترین توجه را به خود اختصاص داده است به سیستم دوپامین mesolimbic گفته می شود که شامل نورون های دوپامین در ناحیه تنگنال واژنی (VTA) متوسط نورون های نوری متوسط در هسته accumbens (NAc، بخشی از striatum شکمی) می شود. این عصب های VTA همچنین بسیاری از مناطق پیش مغز دیگر مانند هیپوکامپ، آمیگدالا و قشر پیشانی (PFC) را مهار می کنند.
این منطقی است که این مکانیسم های اعتیاد ناشی از مواد مخدر را در این حجم برای حافظه برای سه دلایل همپوشانی در نظر بگیریم.6
- اول، تمام سازگاری های ناشی از دارو را می توان به عنوان انواع "حافظه مولکولی یا سلولی" مشاهده کرد: سلول عصبی تحت چنین تغییراتی به عنوان یک نتیجه از مواد مخدر متفاوت است و به همین ترتیب به همان دارو پاسخ می دهد، به داروهای دیگر یا به عنوان یک نتیجه از یک محرک دیگر.
- دوم، جالب است که بسیاری از، شاید بیشتر از انواع تغییراتی که با یک وضعیت اعتیاد همراه بوده اند (به عنوان مثال، رونویسی ژن تغییر یافته، epigenetics، پراکندگی سیناپسی و کل سلولی، و مورفولوژی نورون و مکانیزم های نوروتروف) همچنین در فرم های سنتی "حافظه رفتاری" مانند حافظه فضایی، تهدید ترس، و تهویه عامل، و غیره نیز دخیل هستند.
- سوم، در میان مناطق مغزی که توسط مواد مخدر مورد سوءمصرف قرار می گیرند، کسانی هستند که پایه عصبی کلیدی برای حافظه رفتاری، از جمله هیپوکامپ، آمیگدال و PFC هستند. این همبستگی با تحقق در حال افزایش است که برخی از مهمترین ویژگی های اعتیاد به صورت بالینی دیده می شود (به عنوان مثال، اشتها و رعایت مواد مخدر) منعکس کننده اختلالات در حافظه های حافظه سنتی با خاطرات طولانی مدت از تجربه دارو به عنوان عامل موثر آسیب شناسی اعتیاد است.4,7,8 برعکس ، مناطق پاداش مغز (به عنوان مثال ، VTA و NAc) قدرت حافظه رفتاری را دارند.
این مقاله یک مرور کلی از انواع مختلفی از تغییرات مولکولی و سلولی را که در مدل های حیوانی اعتیاد رخ می دهد در چندین منطقه مغزی رخ می دهد و تمرکز بر تکامل هسته ای است که اکثر اطلاعات در حال حاضر در دسترس است. مهم این است که به طور فزاینده ای ممکن است بعضی از این تغییرات را در معتادان انسانی بر اساس مطالعات مغز پس از قاعدگی به اثبات برسانند. علیرغم اینکه مواد مخدر از سوء استفاده از ساختارهای شیمیایی متمایز استفاده می کنند و بر اهداف متمایز پروتئین عمل می کنند، قابل توجه است که بسیاری از سازگاری های مربوط به اعتیاد برجسته برای بسیاری و در برخی موارد همه، مواد مخدر سوء استفاده و احتمالا به ویژگی های مشترک یک سندرم اعتیاد.4,9 در مقابل، بسیاری از سازگاری های ناشی از مواد مخدر خاص برای یک دارو خاص است و می توانند جنبه های منحصر به فردی از یک اعتبار اعطا شده را در اختیار داشته باشند. ما در اینجا در مورد مواد مخدر محرک و مخدر مورد سوء استفاده قرار می گیریم که در مدل های حیوانی در مقایسه با داروهای دیگر اثرات چشمگیرتری دارد. ما همچنین زمینه های مهم برای تحقیقات آینده را برجسته می کنیم که دانش ما درباره سندرم های اعتیاد را افزایش می دهد و این پیشرفت ها را به آزمایشات و درمان های پیشرفته تشخیصی تبدیل می کند.
مکانیزم های رونویسی و اپی ژنتیک
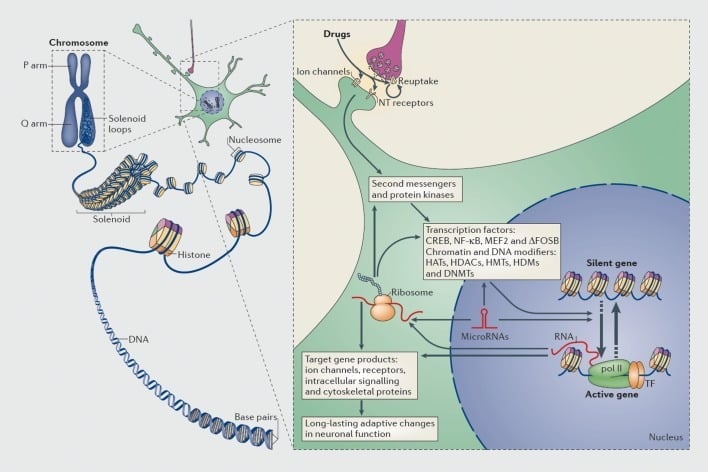
علت این است که معتادان با وجود سالها رعب و وحشت در ریسک ابتلا به عود باقی می مانند، اعتیاد شامل تغییرات ناشی از دارو در مغز می شود که می تواند بسیار پایدار باشد. این باعث شده است که گروه های متعددی تغییراتی را در بیان ژن به عنوان یکی از اجزای مهم فرآیند اعتیاد در نظر بگیرند (شکل 1) بر این اساس، مطالعات ژنهای نامزد یا تحقیقات گسترده ژنوم شامل میکروارگانیسم های DNA و اخیرا RNA-seq (توالی واکنش بالا RNA های بیان شده) ژن های متعددی را شناسایی کرده است که بیان آن در یک منطقه مغز داده شده در مدل های جوانی و پریمات اعتیاد و معتادان به انسان تغییر کرده است (به عنوان مثال، refs 10-17). نمونه هایی از این ژن ها در بخش های بعدی این بررسی مورد بحث قرار گرفته است.
مکانیسم تنظیم رونویسی و اپی ژنتیک توسط داروهای سوء استفاده. در سلول های یوکاریوتی، DNA با پیچاندن اطراف هیستون ها به شکل نوکلئوزوم ها تشکیل می شود که بعدا به شکل کروموزوم (قسمت چپ) سازماندهی می شوند و تشکیل می شوند. تنها با قطع موقت کروماتین فشرده می تواند DNA یک ژن خاص را به دستگاه های رونویسی تبدیل کند. داروهای سوءمصرف از طریق اهداف سیناپسی مانند مکانیسم های مجدد جذب، کانال های یونی و گیرنده های عصبی (NT) برای تغییر آبشارهای سیگنال داخل سلولی (قسمت راست) عمل می کنند. این منجر به فعال شدن یا مهار فاکتورهای رونویسی (TFs) و بسیاری از اهداف هسته ای دیگر، از جمله پروتئین های تنظیم کننده کروماتین می شود (نشان داده شده توسط فلش های ضخیم)؛ مکانیزم های دقیق موجود در تنظیم سیناپسی پروتئین های تنظیم کننده کروماتین پایدار باقی می ماند. این فرایندها در نهایت منجر به سرکوب یا سرکوب ژنهای خاص، از جمله آنهایی که برای RNA های غیرکدینگ مانند میکرو RNA ها هستند؛ بیان تغییری از بعضی از این ژنها به نوبه خود می تواند تنظیم کننده ژن رونویسی باشد. پیشنهاد شده است که برخی از این تغییرات ناشی از دارو در سطح کروماتین بسیار پایدار هستند و بنابراین رفتارهای طولانی مدت که اعتقادات را تعریف می کنند، پایه است. CREB، پروتئین متصل کننده عنصر Response cyclic AMP؛ DNMTs، DNA methyltransferases؛ کلاه، هیستون استیل ترانسفراز؛ HDACs، هیستون deacetylases؛ HDMs، هیستون demethylases؛ HMTs، هیستون متيل ترانسفرازها؛ MEF2، فاکتور تقویتی مزانشیمی 2؛ NF-kB، عامل هسته ای KB؛ پلی II، RNA پلیمراز II. بازسازی شده از ref 44: Robison AJ، Nestler EJ. مکانیزم های رونویسی و اپی ژنتیک اعتیاد. Nat Rev Neurosci. 2011؛ 12: 623-637.
به همین ترتیب، بسیاری از انواع پروتئین رونویسی، پروتئین هایی که به مناطق تنظیم کننده ژن متصل می شوند و درنتیجه باعث افزایش یا کاهش رونویسی این ژن ها می شوند، در میان گذاشتن اثرات درازمدت دارو سوء استفاده بر بیان ژن در مغز. نمونه های برجسته شامل CREB (پروتئین اتصال دهنده واکنش cAMP)، ΔFosB (فاکتور رونویسی خانواده Fos)، NFkB (فاکتور هسته ای kB)، MEF2 (فاکتور MYOCYTE FACTOR-2) و گیرنده های گلوکوکورتیکوئیدها، در میان چندین مورد دیگر.5,10,18-22 درک مسیرهای سیگنالینگ سلولی که از طریق آن داروهای سو of مصرف فاکتور رونویسی مشخصی را در مغز فعال می کنند و ارتباط علنی چنین فعال سازی با ژنهای هدف آن فاکتور رونویسی و جنبه های رفتاری خاص اعتیاد به طور فزاینده ای امکان پذیر شده است (نگاه کنید به شکل 1). این پیشرفت با در نظر گرفتن CREB و ΔFosB نشان داده شده است که بهترین عوامل رونویسی مورد مطالعه در مدل های اعتیاد هستند.
پروتئین اتصال دهنده عنصر پاسخ cAMP
مواد مخدر تحریک کننده و مخدر از سوء استفاده فعال است CREB در چندین منطقه مغزی که برای اعتیاد مهم هستند، از جمله در NAc مهم است.23,24 CREB شناخته شده است در سیستم های دیگر توسط cAMP، Ca فعال می شود2+، و مسیرهای عامل فاکتور رشد25 و هنوز معلوم نیست کدام یک از این ها فعال شدن آن در NAc توسط داروهای سوء استفاده است. نشان داده شده است که فعال سازی دارو CREB در NAc نشان دهنده مکانیسم بازخورد منفی کلاسیک است, به موجب آن CREB به منظور کاهش حساسیت حیوان به اثرات سودمند این داروها (تحمل) و واسطه ای در یک حالت عاطفی منفی در هنگام ترک دارو (وابستگی) عمل می کند..18,26,27 این اثرات اخیرا نشان داده شده است که باعث افزایش مصرف خود دارو و عود می شود، احتمالا از طریق یک فرآیند تقویت منفی.28 به نظر می رسد این اقدامات CREB حاوی هر دو زیرمجموعه های اصلی نورون های کروی متوسط NAc هستند، کسانی که عمدتا D را بیان می کنند1 در مقابل D2 گیرنده های دوپامین.24 Iبه طرز شایان توجه، بخش بزرگی از ادبیات نشان داده است که CREB، در هیپوکامپ و آمیگدال، یک مولکول کلیدی در حافظه رفتاری است.29-31 این نقش گسترده در اعتياد و حافظه رفتاری به احتمال زیاد، این واقعیت را نشان می دهد که نورون ها با تعداد محدودی از مکانیزم های مولکولی که با انطباق با یک محیط دائما در حال تغییر است، نفوذ می کنند.
ژن هدف برای CREB که این فنوتیپ رفتاری را به عهده دارد، از طریق بررسی های ژنوم و همچنین تلاش های بیشتر انتخاب شده است.10,18,32 یک مثال دینورفین پپتید opioid است: القاء تحریک کننده بیان دینورفین در نورون NAc، از طریق CREB، فعال سازی دینورفین گیرنده های K opioid در نورون های دوپامین VTA و در نتیجه باعث انتقال پروتئین dopaminergic به NAc و پاداش را مختل می کند.18 چندین هدف دیگر CREB برای پلاستیک سیناپسی ناشی از مواد مخدر مهم است، همانطور که در زیر بحث شده است. در حالی که CREB نیز توسط محرک ها و مواد مخدر در مناطق مختلف مغز فعال شده است،23,24 کمتر در مورد پیامدهای رفتاری این اثر و ژن های هدف که از طریق آنها رخ می دهد ، شناخته شده است. به همین ترتیب ، کمتر در مورد نقش CREB در میانجیگری اقدامات سایر داروهای سو abuse استفاده شناخته شده است.19
ΔFosB
قرار گرفتن در معرض عمدتا هر گونه سوء مصرف مواد، باعث ایجاد همه عوامل رونویسی خانواده Fos در NAc و چندین مغز دیگر می شود. این القای سریع است اما همچنین بسیار موقت است، با افزایش سطح پروتئین Fos به 8 به 12 ساعت به طور طبیعی. منحصر به فرد در میان این پروتئین خانواده Fos ΔFosB، یک محصول مختلط از ژن FosB، که به سبب ثبات غیرمعمول آن، به تدریج طی دوره ای از آلودگی مواد مکرر تجمع می یابد و پروتئین غالب پروتئین FOS بیان شده در این شرایط می شود.22,33 علاوه بر این، به دلیل این ثبات، سطوح ΔFosB به مدت چند هفته پس از خارج شدن از دارو ادامه می یابد. چنین القایی مزمن ΔFosB برای تقریبا تمام داروهای سوء استفاده نشان داده شده است34 و برای اکثر داروها برای نورونهای NAc نوع Dl انتخابی است.34,35 این نیز وجود دارد در معتادان انسانی نشان داده شده است.35 بخش بزرگی از ادبیات نشان داده است که چنین القاء ΔFosB در D1-نوع نورون های NAc حساسیت حیوان به مواد مخدر و همچنین جوایز طبیعی را افزایش می دهد و باعث تجویز خود دارو می شود ، احتمالاً از طریق فرایند تقویت مثبت (34 به 38 مراجعه کنید). جالب توجه است که القاء دارو ΔFosB در NAc در حیوانات نوجوان بسیار چشمگیر است، زمان آسیب پذیری بیشتر اعتیاد,39 و نشان داده شده است که القا its آن توسط نیکوتین باعث افزایش پاداش کوکائین مانند دروازه مانند نیکوتین می شود.40
همانطور که برای CREB، ژن های هدف متعددی برای ΔFosB در NAc با استفاده از ژن کاندید و روش های ژنوم گسترده شناخته شده است.10,32 در حالی که CREB باعث دیونورفین می شود ، ΔFosB آن را سرکوب می کند ، که به اثرات پاداش ΔFosB کمک می کند.38 هدف دیگر ΔFosB cFos است: به عنوان ΔFosB با تظاهرات مکرر تجمع تجمع می یابد، آن را c-Fos را سرکوب می کند و به سوئی مولکولی کمک می کند که بدینوسیله ΔFosB به صورت انتخابی در حالت درمان دارویی مزمن ایجاد می شود.41 نشان داده شده است که بسیاری از دیگر اهداف ΔFosB توانایی بعضی داروهای سوءاستفاده را برای ایجاد انعطاف پذیری سیناپسی در NAc و تغییرات مرتبط با آن در بافت های دندریتیک نورون های نخاعی متوسط NAc نشان می دهند.
عواقب عملکرد القاء ΔFosB در مناطق مغز دیگر به خوبی درک نشده است، هر چند که القاء آن در قشر اوربیتوفرنتال (OFC) با جزئیات جزئی مورد مطالعه قرار گرفته است. در اینجا، ΔFosB واسطه تحمل است که به اثرات تخریب شناختی کوکائین در طول دوره معاینه مزمن رخ می دهد، و این سازگاری با افزایش خودكارآزمایی كوكائین.42,43
تست های ژنوم گسترده چندین ژن بالقوه احتمالی را پیشنهاد می کنند که میان این اثرات متداول است.42 علی رغم خصوصیات زمانی منحصر به فرد ΔFosB ، و دانش ناشی از آن در مدارهای حافظه سنتی (به عنوان مثال ، هیپوکامپ) ، هنوز در مورد نقش ΔFosB در حافظه رفتاری ، موضوع جالبی برای تحقیقات آینده کشف نشده است.
مکانیزم های اپیزیونیک
در سال های اخیر، مطالعات رونویسی یک مرحله ای به سمت اپی ژنتیک رانده شده است44 (نگاه کنید به شکل 1)، که به طور گسترده ای می تواند به عنوان یک تغییر در بیان ژن تعریف شود که در غیاب تغییر در توالی DNA اتفاق می افتد. مکانیسم های اپیزیونیک بسته بندی DNA را در یک هسته سلولی کنترل می کند، از طریق تعاملات آن با هیستون ها و بسیاری از انواع دیگر پروتئین های هسته ای، که همگی شامل کروماتین می شوند. بیان ژن توسط دولت این بسته توسط تغییرات کووالانستی هیستونها، پروتئین های دیگر و خود DNA کنترل می شود. همانطور که فقط برخی از نمونه ها، استیلینگ هیستون ها تمایل به ترویج فعال سازی ژن دارد، متیلاسیون هیستون ها می تواند به تقویت فعال سازی ژن یا سرکوب بستگی به بقایای Lys تحت این اصلاح داشته باشد و متیلاسیون DNA عموما با سرکوب ژن همراه است، به عنوان مثال، 5-hydroxymethylation) ممکن است با فعال سازی ژن مرتبط باشد.
Epigenetics مکانیکی جذاب است، زیرا در سایر سیستم ها، برای مثال، زیست شناسی در حال توسعه و سرطان، برخی از تغییرات اپی ژنتیکی می توانند دائمی باشند. به این دلیل، اپی ژنتیک در مدل یادگیری و حافظه (به عنوان مثال، refs 45-48) و همچنین در اعتیاد دنبال شده است.44,49 در هر دو سیستم تغییرات عمیق در استیلاسیون و متیلاسیون هیستون و در متیلاسیون DNA گزارش شده است. به عنوان یک مثال، هیستون متیل ترانسفراز، G9a، در هر دو حافظه دخیل است50 و اعتیاد51,52 در مدل های اعتیاد بیان G9a غلط است در NAc در پاسخ به داروهای محرک یا مخدر مصرف سوء مصرف مواد و غیرهنشان داده شده است که اثرات مفید این داروها را افزایش داده است.51,52 جالب توجه است، سرکوب کوکائین G9a توسط ΔFosB مت starp می شود. G9a کاتالیز دی متیلسیون Lys9 از هیستون H3 (H3K9me2)، یک واسطه عمده سرکوب ژن. Chip chip یا ChIP-seq (کروماتین ایزوپروپانیس پس از آن به ترتیب توسط تراشه های پروموتور یا توالی فرکانس بالا) برای بدست آوردن نقشه های ژنوم ژن در NAc که نشان دهنده تغییر H3K9me2 پس از قرار گرفتن در معرض محرک یا مخدر است استفاده شده است.32,52,53 با همپوشانی این لیست ژن با لیست ژنوم گسترده ای از تغییرات بیان ژن، و با نقشه های ژنوم گسترده از بسیاری از اشکال دیگر تغییرات اپی ژنتیک (به عنوان مثال، اتصال ΔFosB، اتصال CREB، دیگر تغییرات هیستون و غیره)،32,53 باید مجموعه ای از ژن های به طور کامل کامل را شناسایی کرد که توسط مواد مخدر مورد سوءاستفاده قرار می گیرند و برای درک مکانیسم های اپی ژنتیک زیرمجموعه.
شکل دیگری از تنظیمات اپی ژنتیک که در حافظه و اعتیاد دخیل هستند تولید نسل microRNAs است. این RNA های کوچک و غیر کدومی به مناطق مکمل از mRNA ها متصل می شوند و درنتیجه آنها را از بین می برد یا باعث تخریب آنها می شوند. حذف Argonaut، یک پروتئین حیاتی برای پردازش miRNA ها، واکنش های رفتاری کوکائین را تغییر می دهد و اثرات متفاوتی را برای نورون های کروی متوسط D1 و D2 مشاهده می کند.54 همچنین نشان داده شده است که چندین miRNA اختصاصی به وسیله قرار گرفتن در معرض دارو تنظیم می شوند و به نوبه خود برای تأثیر رفتارهای رفتاری به داروها (به عنوان مثال، refs 55,56) تنظیم می شود. این در مطالعات آینده هیجان انگیز خواهد بود تا اهداف mRNA این miRNA ها را شناسایی کند و مشخص کند چگونه بر روند اعتیاد تأثیر می گذارد.
پلاستیک سیناپتیس
همان گونه کلی نوع تغییرات سیناپسی در سیناپس های گلوتاماترگیک که در حافظه رفتاری در هیپوکامپ و آمیگدال دخیل هستند (به مقالات دیگر در این موضوع مراجعه کنید)، به طور مشابه نشان داده شده است که در مناطق پاداش مغز در مدل های اعتیاد رخ داده است و در میانجیگری مهم هستند فرایند اعتیاد.57,58 چنین پلاستیک سیناپسی ناشی از مواد مخدر در مناطق مختلف مغز توصیف شده است، با این حال، ما در اینجا تمرکز داریم بر روی NAc که بیشتر تحقیقات تا به امروز متمرکز شده است (شکل 2).
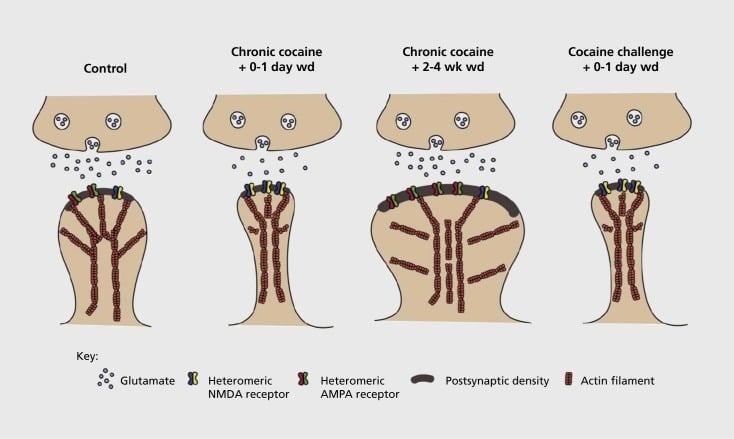
مدل وابستگی سیناپسی و ساختاری پلاستیک در هسته accumbens (NAc). قرار گرفتن در معرض مکرر کوکائین باعث تغییر زمان گیرنده ی گیرنده های گلوتامات α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) و N-methyl-D-aspartic acid (NMDA) در محیط NAc می شود. سیناپس نرون مینور (MSN) و همچنین تغییرات ساختاری در ستون فقرات NAc MSNs که با اشکال متمایزی از پلاستیک سیناپسی مرتبط است. به عنوان مثال، کوکائین مزمن ناشی از بیان سطح گیرنده های NMDA، شکل گیری سیناپس خاموش و افسردگی طولانی مدت (محدودیت) در مراحل زود هنگام زودرس است. در طول طولانی شدن خروج (wd)، این تغییرات سیناپسی معکوس می شود، در نتیجه افزایش بیان کننده گیرنده های AMPA سطح، تقویت سیناپس به ستون فقرات شکل قارچ و potentiation طولانی مدت (LTP) می شود. این اثرات به سرعت در معرض دوز چالش کوکائین است که منجر به بازسازی ستون فقرات در ستون فقرات و افسردگی قدرت سیناپسی می شود.
آزمایش های اولیه نشان داد که قرار گرفتن در معرض مکرر داروهای تحریک کننده سوءمصرف باعث ایجاد افسردگی شدید (مانند افسردگی طولانی مدت) در سیناپس گلوتاماترگیک در NAc می شود.59 با این حال، کارهای اخیر اخیر نشان داده است که چنین انعطاف پذیری به شدت وابسته به زمان است، با محدودیت زمانی که اوایل پس از آخرین قرار گرفتن در معرض کوکائین تبدیل به بیشتر از حالت LTP (potentiation طولانی مدت) پس از زمان طولانی تر از زمان برداشت.60,61 این کار که تا به امروز انجام شده است، عمدتا با استفاده از محقق انجام شده به عنوان مخالفت با داروهای خود اداره شده است، نیاز به بررسی بیشتر سیستماتیک در مدل های خودآموزی دارد که فرمهای پلاستیسیت سیناپسی را در سیناپس گلوتاماترگیک در NAc در طول زمان دقیق از کسب خود مدیریت به تعمیر و نگهداری آن، از طریق زمان های مختلف از عقب نشینی و انقراض، و در پاسخ به انگیزه های بازگشت عود. کار تا به امروز نیز برخی از مکانیزم های مولکولی که به این پلاستیک سیناپسی ناشی از مواد مخدر کمک می کند، از جمله قاچاق گیرنده های AMPA به سیناپس، شاید بخشی از طریق CaMKII (Ca2+/ پروتئین کیناز II وابسته به کالودولین II) فسفریلینگ برخی از واحدهای گیرنده AMPA و همچنین بیان تغییرات واحدهای گیرنده AMPA (به عنوان مثال 60,62-65، ارقام 2 و 3). نقش CREB و ΔFosB در این پدیده ها و نیز تغییرات مرتبط در مورفولوژی سیناپس گلوتاماترگیک (به زیر) اشاره شده است. به عنوان مثال، GluAl یک هدف برای CREB در NAc است، جایی که GluA2 و CaMKII هر دو اهداف ΔFosB هستند، در این منطقه مغز .35,36,66,67 حرکت به جلو، مهم است که ارتباط سازگاری خاص با تغییرات وابسته به زمان در عملکرد سیناپسی و ویژگی های رفتاری اعتیاد داشته باشیم.
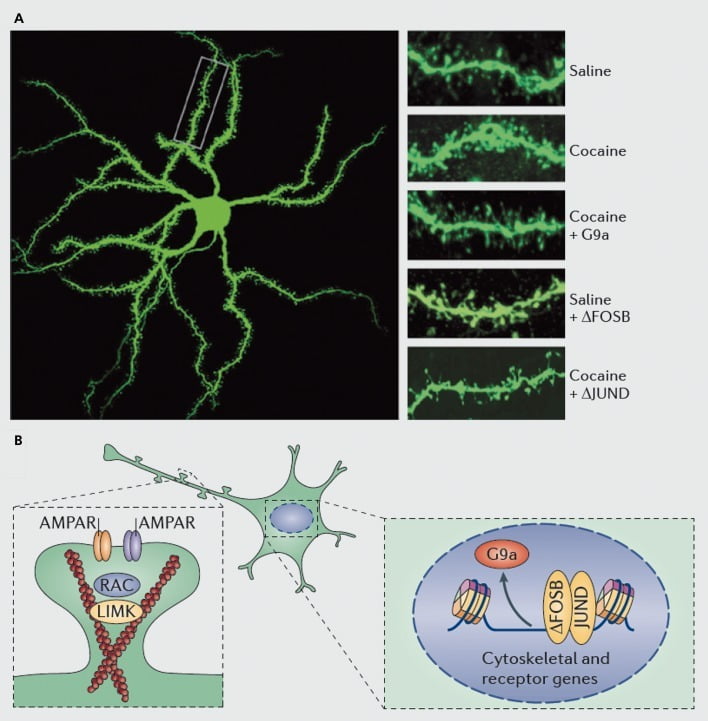
مکانیسم های مولکولی زمینه ساز القای کوکائین ستون فقرات دندریتیک در هسته های هسته ای (NAc) سلول های عصبی خاردار متوسط. الف) افزایش ناشی از کوکائین در تعداد ستون فقرات دندریتیک را نشان می دهد که می تواند با بیان زیاد ویروسی G9a یا JunD (آنتاگونیست رونویسی با واسطه AP1) مسدود شود ، یا با بیان بیش از حد ویروسی FosB تقلید شود. ب) نشان داده شده است که تنظیم قاچاق گیرنده های AMPA (AMPAR) و اسکلت سلولی اکتین (چپ) ، و همچنین تنظیم رونویسی از گیرنده های گلوتامات و پروتئین های تنظیم کننده اکتین (به عنوان مثال ، با واسطه از طریق ΔFosB ، سمت راست) نقش مهمی دارند در واسطه تنظیم کوکائین از NAc تراکم ستون فقرات دندریتیک - سایپرز ، باشگاه دانش دامنه کیناز UMK ، LIM ؛ RAC ، بستر سم بوتولینوم مربوط به Ras C.
ابزار تجربی جدید، برای اولین بار با دقت بالا تعریف می شود که کدامیک از مدارهای خاص، این اشکال انعطاف پذیری سیناپسی را نشان می دهند و اینکه چه اختلالات رفتاری آنها میانجیگر هستند. به عنوان مثال، tاو پوسته و زیرمجموعه های اصلی NAc تفاوت های مختلفی در پلاستیک سیناپسی ناشی از مواد مخدر نشان می دهد، همانطور که D1- و D2-نوع متوسط نرون های خاردار متوسط در هر زیرمجموعه.60,63,64,67 به همین ترتیب، آزمایشات optogenetic بینش جدیدی را در مورد مشارکت یک فرم خاص از پلاستیک سیناپسی (به عنوان مثال، LTD) در جمعیت خاص از سیناپس گلوتاماترگیک در NAc، ارائه شده است، به عنوان مثال، کسانی که ناشی از PFC medial در مقابل amygdala مقعدی در مقابل ساب سکتوم شکمی (خروجی عمده از هیپوکامپ)68-70 در نهایت ، لازم است سازگاری های مولکولی ناشی از دارو در هر یک از این سلول های عصبی آوران با انطباق های خاص سیناپس که در دندریت های پس سیناپسی آنها رخ می دهد ، پوشانده شود تا درک کاملی از نحوه تغییر داروهای سو drugs استفاده در مدار مغز برای ایجاد جنبه های خاص حالت معتاد این تلاش نیاز به درک بیشتر از انعطاف پذیری ناشی از دارو در سیناپس های مهاری در همان مناطق مغزی دارد ، منطقه ای که تا به امروز بسیار کم مورد توجه قرار گرفته است.65
انعطاف پذیری کامل سلول
در حالی که اکثر تحقیقات مربوط به تغییرات نوروفیزیولوژیک در نورون ها در پدیده سوء مصرف مواد، به عنوان در یادگیری و پدیده حافظه، بر روی انعطاف پذیری سیناپسی متمرکز شده است، شواهد افزایشی در مورد اهمیت پلاستیک کامل سلول نیز وجود دارد. انعطاف پذیری کامل سلول، همچنین به عنوان پلاستیسیته خانه شناخته شده است71 شامل تغییرات در تحریک پذیری ذاتی یک کل سلول عصبی به شیوه ای است که خاصه سیناپس نیست. با توجه به این که ویژگی های خاصی از اعتیاد به مواد مخدر باعث افزایش یا کاهش حساسیت به یک داروی می شود، منطقی است که افزایش یا کاهش قابلیت تحریک پذیری الکتریکی برخی سلول های عصبی در این سازگاری های رفتاری.5
La بهترین مثال ثابت شده از پلاستیسیته کامل سلول به دارو سوء استفاده، توانایی opiates مزمن برای افزایش تحریک پذیری ذاتی نورون های نئودرنرژیک locus coeruleus (LC).72 این افزایش تحریک پذیری از طریق CREB و القاء ایزوفرم های خاصی از آدنیلیل سیکلوس است که باعث افزایش شلیک نورون های LC می شود، شاید از طریق ایجاد کانال های Na +.72-75 این هیجانات باورنکردنی نورون های LC نشان دهنده یک مکانیزم کلاسیک تحمل و وابستگی است و برخی از علائم و نشانه های خروج از مواد مخدر را در بر می گیرد. جالب توجه است، CREB یک شکل مشابه از پلاستیک کامل سلول را در نورونهای کروی متوسط NAc متمایز می کند، که در معرض ابتلا به مواد مخدر سوء استفاده از طریق CREB نیز از بین می رود.76 بنابراین در پژوهش های آینده، برای درک چگونگی پلاستیک سیناپسی سیناپس گلوتاماترگیک بر روی نورون های نازک متوسط NAc،65,66 summates با CREB-mediated غربالگری درونی این نورون76 برای کنترل ویژگی های رفتاری اعتیاد.
نمونه دیگری از پلاستیک کامل سلول در مدل های اعتقادی، افزایش قابل توجهی از نورون های دوپامین VTA است که پس از قرار گرفتن در معرض مواد منفجره مزمنوشکل 4).77,78 این سازگاری که با تغییرات مورفولوژیکی در این سلول های عصبی ارتباط دارد (نگاه کنید به بخش بعدی)، توسط CREB متاثر نیست، بلکه به وسیله تنظیم آبشارهای سیگنال نوروتروف، به صورت زیر شرح داده شده است.

مدل کاري سازگاري ناشي از مورفين مزمن در نورونهاي دوپامين منطقه Ventral Temental Area (VTA). مورفین مزمن باعث کاهش اندازه سمی VTA دوپامین (DA) می شود، اما تحریک پذیری نورون را افزایش می دهد، در حالیکه انتقال دوپامین به nucleus accumbens کاهش می یابد. اثر خالص مورفین یک مسیر پاداش کمتر پاسخگو است، یعنی تحمل پاداش. کمینه شدن سیگنال IRS2-AKT در VTA، باعث کاهش اثرات مورفین مزمن بر اندازه سما و تحریک پذیری الکتریکی می شود. اثر بر تحریک پذیری از طریق کاهش اسید γ-آمینوبوتیریک (GABA) A و سرکوب بیان کانال K. کاهش تنظیم فعالیت مورفین فعالیت mTORC2 در VTA برای این سازگاری های مورفولوژیکی و فیزیولوژیک ناشی از مورفین و نیز تحمل پاداش بسیار مهم است. در مقایسه با mT0RC2، مورفین مزمن فعالیت mTORCI را افزایش می دهد که بر سازگاری ناشی از مورفین تاثیر نمی گذارد. BDNF، فاکتور نوروتروفی حاصل از مغز؛ IRS، ماده گیرنده انسولین؛ mTORC، mTOR complex؛ AKT، پروتئین کیناز B تولید شده از ref 77
پلاستیکی مورفولوژی و مکانیسم های نوروتروفیک
شواهد افزایش یافته، بسیاری از مطالعات در مورد نورون های قشر هیپوکامپ و مغز، نشان داده است که تغییرات در پلاستیک سیناپسی با تغییرات مورفولوژیکی در سیناپس مرتبط است. به عنوان مثال، محدود کردن و تولید سیناپس های خاموش با تشکیل ستون های دندریتیک نازک یا قاعده همراه است، در حالی که LTP با بزرگترها، ستون های قارچی شکل تشکیل شده است.79,80 بنابراین جالب است که زمینه سو abuse مصرف مواد مخدر بیش از 15 سال بر تغییرات ناشی از دارو در ستون فقرات دندریتیک تمرکز کرده است. قرار گرفتن در معرض مزمن داروهای تحریک کننده سوءمصرفه تراکم ستون فقرات دندریتیک نورونهای نوری متوسط NAc را افزایش می دهد، تغییری که برای نورونهای Dl غالب است.67,81,82 القاء ستون فقرات به طور عمده با پاسخ های رفتاری حساس به این داروها ارتباط دارد، هرچند برخی شواهد با این دیدگاه مخالف هستند.
همانطور که با مطالعات پلاستیسیته سیناپسی، به منظور سیستمیک تغییرات در ستونهای دندریتیکی که در طول دوره تزریق دارو، خروج و عود بیماری رخ می دهد، نیاز به کار بیشتر است. Sدر حال حاضر، شامل داروهای تحقیق کننده و خودمراقبت، نشان می دهد که تغییرات ستون فقرات بسیار متفاوت در نقاط مختلف زمان خروج و در ناحیه پوسته در مقابل زیرمجموعه های اصلی.83-86 همچنین تعیین مکانیزم های دقیق مولکولی که کوکائین و یا یک محرک دیگر باعث ایجاد اثرات متفاوتی وابسته به زمان و سلول می شوند، مهم است. ΔFosB نشان داده شده است که هر دو لازم و کافی برای القاء ستون های نابالغ در NAl نوع نورون Dl.35,51,67 چنین مقرراتی با هماهنگی با کوکائین و تنظیم ΔFosB از چندین پروتئین شناخته شده برای کنترل سازماندهی مجدد سیتو اسکلت اکتین اتفاق می افتد. همانطور که فقط یک مثال، تنظیم رونویسی چندین فاکتور تبادل گوانین نوکلئوتیدی و پروتئین فعال کننده GTPase، Rac1 کوچک، GTPase کوچک، برای کاهش گذرا در فعالیت در پاسخ به هر گونه قرار گرفتن در معرض هر کوکائین، و از جمله کاهش pulsatile در فعالیت Rac1، با استفاده از کنترل optogenetic از Rac1، برای ایجاد القاء ستون فقرات نابالغ.87 این اثرات راسل احتمالا از طریق کنترل کافیلین و دیگر پروتئین های تنظیم کننده اکتین اتفاق می افتد که نشان می دهد که تنظیم کننده کوکائین از رشد ستون فقرات است.87,88 با این حال ، تأکید بر این نکته مهم است که این تنها یک مسیر در تنظیم ستون فقرات نابالغ کوکائین است ، زیرا چندین پروتئین دیگر نیز نقش اساسی دارند ، از جمله CDK5 (کیناز 5 وابسته به سیکلین) ، CaMKII ، NFkB ، MEF2 ، CREB ، G9a و DNMT3 (DNA متیل ترانسف 3a را پاک می کند) ، به چند مورد.20,21,35,51,67,89,90 جالب توجه است که تعدیل کوکائین چندین از این ژنها، از جمله القاء CDK5، CaMKII و NFkB، و سرکوب G9a نیز از طریق ΔFosB متمرکز می شود.20,35,51,91
به طرز شگفتانگیزی، داروهای سوء مصرف مواد مخدر، اثر متفاوتی را اعمال می کنند و تراکم ستون فقرات دندریتیک نورون های نخاعی متوسط NAc را کاهش می دهند..81 کمی در مورد پیامدهای رفتاری این سازگاری و سازوکارهای مولکولی موجود در آن، کم است. این پدیده است، با این حال، شگفت آور است، با توجه به اینکه CREB و ΔFosB بوسیله هر دو محرک و opiates القا می شوند و هر دو در تحریک مبتنی بر القاء از NAc تراکم ستون فقرات دندریتیک. این مسئله مسأله ای را مطرح می کند که چگونه مواد افیونی، چگالی ستون فقرات را کاهش می دهد، علیرغم القاء این عوامل.
شکل اصلی دیگری از پلاستیک مورفولوژیکی که در مدل های سوء مصرف مواد دیده می شود، کاهش فیزیکی در اندازه سم سلولی نورون های دوپامین VTA ناشی از تزریق مزمن مزمن است.77,92,93 سازگاری مشابه در پاسخ به کانابینوئید ها رخ می دهد.94 این انقباض نورونهای دوپامین VTA، که با خودکشی اپیدمی رخ می دهد93 و در معتادان هروئین انسان مستقیما مورد بررسی قرار گرفت،77 به نظر می رسد به تحریم پاداش می پردازد و با کاهش حجم آزاد شدن دوپامین در NAc ارتباط دارد. شواهد قابل ملاحظه ای در حال حاضر نشان می دهد که این کاهش در اندازه سومی سلولی به وسیله مهار افیون از بیان نوروپتیک فاکتور مغز (BDNF) در این نورون ها متاثر می شود. ما به طور مستقیم از برداشتن حمایت از BDNF ناشی از مواد مخدر و کاهش انقباض عصبی VTA برای کاهش فعالیت آبشارهای سیگنال BDNF پایین در نورون های دوپامین VTA، به طور خاص کاهش فعالیت IRS2 (گیرنده تزریق انسولین 2)، AKT (سرین ترئونین کیناز) و TORC2 (هدف از rapamycin-2، که به rapamycin حساس نیستند).77,93 همانطور که قبلا اشاره شد، ما نیز این کاهش تنظیم BDNF سیگنالینگ را به طور مستقیم به افزایش تحریک پذیری مرتبط با مورفین در این نورون ها مرتبط کرده ایم.77,78 در واقع، میزان کاهش سم سلول و افزایش تحریک پذیری به شدت متصل است، زیرا القاء یکی به سمت دیگر و بالعکس است. این کنترل بر تحریک پذیری سلولی شامل سرکوب K است+ کانال ها و GABAA در این نورونها جریان دارد.
این نقش برای BDNF در کنترل واکنش های مورفین در سطح VTA در مقایسه با درگیری های بسیار متفاوتی در فعالیت کوکائین و سایر محرک ها است. تحریک کننده باعث ایجاد سیگنالینگ BDNF به NAc می شود، اثر ناشی از افزایش سنتز محلی BDNF و افزایش آزاد شدن آن از چندین ناحیه جانبی.95 علاوه بر این، افزایش سیگنالینگ BDNF در NAc، اما نه در VTA، اثبات شده است که اثرات رفتاری این داروها شامل خودکارش را افزایش می دهد.95,96 تنظیم مخالف سیگنالینگ BDNF در مسیر VTA-NAc توسط مواد افیونی در مقابل مواد محرک این احتمال را ایجاد می کند که چنین اختلافاتی باعث تنظیم واسطه دارو در ستون فقرات دندریتیک NAc شود ، امکانی که اکنون تحت بررسی است.
دستورالعمل های آینده
روایت فوق بر پیشرفت های فوق العاده ای که در درک سازگاری های مولکولی و سلولی رخ داده است که در مناطق پاداش مغز رخ می دهد در واکنش به مواجهه مکرر با مواد مخدر سوء استفاده و در ارتباط سازگاری فرد با ویژگی های رفتاری برخی از سندرم های اعتیاد در مدل های حیوانات . با وجود این پیشرفت ها، سوالات اصلی همچنان ادامه دارد. اکثر دانش موجود ما بر روی VTA و NAc تمرکز می کنند و اطلاعات بسیار کمتری در مورد دیگر مناطق مغزی لمبی مغز وجود دارد که برای اعتیاد به مواد مخدر نیز بسیار مهم هستند. علاوه بر این، تمام تظاهرات تجربی نقش علمی انطباق سلولی مولکولی در رفتار مربوط به مواد مخدر، انطباق فردی را در یک زمان دستکاری کرده است. برای مقابله با تعدیل های متعدد در همان زمان، به وضوح بسیار دشوار است، اما این نیز ضروری است، زیرا ما می دانیم که داروها تعداد زیادی از انواع مختلف تغییرات را حتی درون سلول های عصبی ایجاد می کنند که احتمالا در شیوه های پیچیده ای برای تاثیر گذار بر آنها تاثیر می گذارد. چنین رویکرد زیست شناسی سیستم، برای نهایتا شکستن پایه های زیست شناختی اعتیاد بسیار مهم خواهد بود. در نهایت، تلاش برای درک مکانیسم های مولکولی سلولی خاطرات مربوط به اعتیاد، خود را در نقطه ای قرار می گیرند که در آن تمام تلاش های دیگر برای درک مبانی بیولوژیکی حافظه رفتاری در حال مبارزه است: توانایی ما در ارتباط پدیده های بیولوژیکی با حافظه پیچیده رفتاری بسیار دشوار است. غلبه بر این تقسیم احتمالا بزرگترین چالش در علوم اعصاب است.
اختصارات و اختصارات انتخاب شده
- ناک
- هسته accumbens
- CREB
- پروتئین اتصال دهنده عنصر پاسخ cAMP
- ΔFosB
- فاکتور رونویسی خانواده Fos
- VTA
- منطقه تکتونال شکمی
- AMPA
- α-آمینو-3-هیدروکسی-5-methyl-4-isoxazolepropionic acid
- محدود
- افسردگی طولانی مدت
- LTP
- پروستات طولانی مدت
- BDNF
- فاکتور نوروپاتی فکری مغز
- NKkB
- فاکتور هسته ای kB
مراجع