COMMENTS: As later studies will reveal DeltaFosB is the common molecular switch for both drug and behavioral addictions. It’s a transcription factor which means it affects what genes are turned on or off. As stated elsewhere, addictive drugs only hijack normal mechanisms. That’s why it’s silly to suggest that behavioral addictions cannot exist.
Proc Natl Acad Sci U S A. 2001 September 25; 98(20): 11042–11046.
doi: 10.1073/pnas.191352698.
Eric J. Nestler*, Michel Barrot, and David W. Self
Department of Psychiatry and Center for Basic Neuroscience, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9070
Abstract
The longevity of some of the behavioral abnormalities that characterize drug addiction has suggested that regulation of neural gene expression may be involved in the process by which drugs of abuse cause a state of addiction. Increasing evidence suggests that the transcription factor ΔFosB represents one mechanism by which drugs of abuse produce relatively stable changes in the brain that contribute to the addiction phenotype. ΔFosB, a member of the Fos family of transcription factors, accumulates within a subset of neurons of the nucleus accumbens and dorsal striatum (brain regions important for addiction) after repeated administration of many kinds of drugs of abuse. Similar accumulation of ΔFosB occurs after compulsive running, which suggests that ΔFosB may accumulate in response to many types of compulsive behaviors. Importantly, ΔFosB persists in neurons for relatively long periods of time because of its extraordinary stability. Therefore, ΔFosB represents a molecular mechanism that could initiate and then sustain changes in gene expression that persist long after drug exposure ceases. Studies in inducible transgenic mice that overexpress either ΔFosB or a dominant negative inhibitor of the protein provide direct evidence that ΔFosB causes increased sensitivity to the behavioral effects of drugs of abuse and, possibly, increased drug seeking behavior. This work supports the view that ΔFosB functions as a type of sustained “molecular switch” that gradually converts acute drug responses into relatively stable adaptations that contribute to the long-term neural and behavioral plasticity that underlies addiction.
Addiction research is focused on understanding the complex ways in which drugs of abuse change the brain to cause behavioral abnormalities that characterize addiction. One of the critical challenges in the field is to identify relatively stable drug-induced changes in the brain to account for those behavioral abnormalities that are particularly long-lived. For example, a human addict may be at increased risk for relapse even after years of abstinence.
The stability of these behavioral abnormalities has led to the suggestion that they may be mediated, at least in part, through changes in gene expression (1–3). According to this view, repeated exposure to a drug of abuse repeatedly perturbs transmission at particular synapses in the brain that are sensitive to the drug. Such perturbations eventually signal via intracellular messenger cascades to the nucleus, where they first initiate and then maintain changes in the expression of specific genes. A primary mechanism through which signal transduction pathways influence gene expression is the regulation of transcription factors, proteins that bind to regulatory regions of genes and modify their transcription.
One goal of addiction research, therefore, has been to identify transcription factors that are altered in brain regions implicated in addiction after chronic administration of drugs of abuse. Several such transcription factors have been identified over the past decade (1–6). The focus of this review is on one particular transcription factor called ΔFosB.
Induction of ΔFosB by Drugs of Abuse
ΔFosB, encoded by the fosB gene, is a member of the Fos family of transcription factors, which also include c-Fos, FosB, Fra1, and Fra2 (7). These Fos family proteins heterodimerize with Jun family proteins (c-Jun, JunB, or JunD) to form active AP-1 (activator protein-1) transcription factors that bind to AP-1 sites (consensus sequence: TGAC/GTCA) present in the promoters of certain genes to regulate their transcription.
These Fos family proteins are induced rapidly and transiently in specific brain regions after acute administration of many drugs of abuse (Fig. 1) (8–11). Prominent regions are the nucleus accumbens and dorsal striatum, which are important mediators of behavioral responses to the drugs, in particular, their rewarding and locomotor-activating effects (12, 13). These proteins return to basal levels within hours of drug administration.
Figure 1
Scheme showing the gradual accumulation of ΔFosB versus the rapid and transient induction of other Fos family proteins in response to drugs of abuse. (A) The autoradiogram illustrates the differential induction of these various proteins by acute stimulation (1–2 hr after a single drug exposure) versus chronic stimulation (1 day after repeated drug exposure). (B) Several waves of Fos-like proteins [comprised of c-Fos (52- to 58-kDa isoforms), FosB (46- to 50-kDa isoforms), ΔFosB (33-kDa isoform), and Fra1 or Fra2 (40 kDa)] are induced in nucleus accumbens and dorsal striatal neurons by acute administration of a drug of abuse. Also induced are biochemically modified isoforms of ΔFosB (35–37 kDa); they, too, are induced (although at low levels) after acute drug administration, but persist in brain for long periods because of their stability. (C) With repeated (e.g., twice daily) drug administration, each acute stimulus induces a low level of the stable ΔFosB isoforms, which is indicated by the lower set of overlapping lines that indicate ΔFosB induced by each acute stimulus. The result is a gradual increase in the total levels of ΔFosB with repeated stimuli during a course of chronic treatment, which is indicated by the increasing stepped line in the graph.
Very different responses are seen after chronic administration of drugs of abuse (Fig. 1). Biochemically modified isoforms of ΔFosB (molecular mass 35–37 kDa) accumulate within the same brain regions after repeated drug exposure, whereas all other Fos family members show tolerance (that is, reduced induction compared with initial drug exposures). Such accumulation of ΔFosB has been observed for cocaine, morphine, amphetamine, alcohol, nicotine, and phencyclidine (11, 14–18). There is some evidence that this induction is selective for the dynorphin/substance P-containing subset of medium spiny neurons located in these brain regions (15, 17), although more work is needed to establish this with certainty. The 35- to 37-kDa isoforms of ΔFosB dimerize predominantly with JunD to form an active and long-lasting AP-1 complex within these brain regions (19, 20). These ΔFosB isoforms accumulate with chronic drug exposure because of their extraordinarily long half-lives (21), and therefore persist in the neurons for at least several weeks after cessation of drug administration. It is interesting to note that these ΔFosB isoforms are highly stable products of an immediate early gene (fosB). The stability of the ΔFosB isoforms provides a novel molecular mechanism by which drug-induced changes in gene expression can persist despite relatively long periods of drug withdrawal.
Although the nucleus accumbens plays a critical role in the rewarding effects of drugs of abuse, it is believed to function normally by regulating responses to natural reinforcers, such as food, drink, sex, and social interactions (12, 13). As a result, there is considerable interest in a possible role of this brain region in other compulsive behaviors (e.g., pathological overeating, gambling, exercise, etc.). For this reason, we examined whether ΔFosB is regulated in an animal model of compulsive running. Indeed, the stable 35- to 37-kDa isoforms of ΔFosB are induced selectively within the nucleus accumbens in rats that show compulsive running behavior.†
Biochemical Identity of Stable ΔFosB Isoforms
As mentioned above, the ΔFosB isoforms that accumulate after chronic administration of a drug of abuse or compulsive running show a molecular mass of 35–37 kDa. They can be differentiated from the 33-kDa isoform of ΔFosB that is induced rapidly but transiently after a single drug exposure (Fig. 1) (14, 19, 22). Current evidence suggests that the 33-kDa isoform is the native form of the protein, which is altered to form the more stable 35- to 37-kDa products (19, 21). However, the nature of the biochemical modification that converts the unstable 33-kDa isoform into the stable 35- to 37-kDa isoforms has remained obscure. It has been speculated that phosphorylation may be responsible (11). For example, induction of ΔFosB is attenuated in mice lacking DARPP-32, a striatal-enriched protein (23, 24). Because DARPP-32 regulates the catalytic activity of protein phosphatase-1 and protein kinase A (25, 26), the requirement for this protein for the normal accumulation of the stable ΔFosB isoforms suggests a possible role for phosphorylation in generation of these stable products.
Role of ΔFosB in Behavioral Plasticity to Drugs of Abuse
Insight into the role of ΔFosB in drug addiction has come largely from the study of transgenic mice in which ΔFosB can be induced selectively within the nucleus accumbens and other striatal regions of adult animals (27, 28). Importantly, these mice overexpress ΔFosB selectively in the dynorphin/substance P-containing medium spiny neurons, where the drugs are believed to induce the protein. The behavioral phenotype of the ΔFosB-overexpressing mice, which in many ways resembles animals after chronic drug exposure, is summarized in Table 1. The mice show augmented locomotor responses to cocaine after acute and chronic administration (28). They also show enhanced sensitivity to the rewarding effects of cocaine and morphine in place-conditioning assays (11, 28) and will self-administer lower doses of cocaine than littermates that do not overexpress ΔFosB.‡ In contrast, these animals show normal conditioned locomotor sensitization to cocaine and normal spatial learning in the Morris water maze (28). These data indicate that ΔFosB increases an animal’s sensitivity to cocaine and perhaps other drugs of abuse and may represent a mechanism for relatively prolonged sensitization to the drugs.
 striatum
striatum
| Increased locomotor activation in response to acute and repeated cocaine administration. |
| Increased rewarding responses to cocaine and morphine in place-conditioning assays. |
| Increased self-administration of low doses of cocaine. |
| Increased motivation for cocaine in progressive ratio assays. |
| Increased anxiolytic responses to alcohol. |
| Increased compulsive running behavior. |
Based on data in refs. 28 and 29.†‡§¶
Behavioral plasticity mediated by ΔFosB in nucleus accumbens-dorsal striatum
In addition, there is preliminary evidence that the effects of ΔFosB may extend well beyond a regulation of drug sensitivity per se to more complex behaviors related to the addiction process. Mice expressing ΔFosB work harder to self-administer cocaine in progressive ratio self-administration assays, suggesting that ΔFosB may sensitize animals to the incentive motivational properties of cocaine and thereby lead to a propensity for relapse after drug withdrawal.‡ ΔFosB-expressing mice also show enhanced anxiolytic effects of alcohol,§ a phenotype that has been associated with increased alcohol intake in humans. Together, these early findings suggest that ΔFosB, in addition to increasing sensitivity to drugs of abuse, produces qualitative changes in behavior that promote drug-seeking behavior. Thus, ΔFosB may function as a sustained “molecular switch” that helps initiate and then maintain crucial aspects of the addicted state. An important question under current investigation is whether ΔFosB accumulation during drug exposure promotes drug-seeking behavior after extended withdrawal periods, even after ΔFosB levels have normalized (see below).
Adult mice that overexpress ΔFosB selectively within the nucleus accumbens and dorsal striatum also exhibit greater compulsive running compared with control littermates.† These observations raise the interesting possibility that ΔFosB accumulation within these neurons serves a more general role in the formation and maintenance of habit memories and compulsive behaviors, perhaps by reinforcing the efficacy of neural circuits in which those neurons function.
ΔFosB accumulates in certain brain regions outside the nucleus accumbens and dorsal striatum after chronic exposure to cocaine. Prominent among these regions are the amygdala and medial prefrontal cortex (15). A major goal of current research is to understand the contributions of ΔFosB induction in these regions to the addiction phenotype.
Earlier work on fosB knockout mice revealed that these animals fail to develop sensitization to the locomotor effects of cocaine, which is consistent with the findings of the ΔFosB-overexpressing mice mentioned above (22). However, the fosB mutants showed enhanced sensitivity to cocaine’s acute effects, which is inconsistent with these other findings. Interpretation of findings with the fosB mutants, though, is complicated by the fact that these animals lack not only ΔFosB, but full-length FosB as well. Moreover, the mutants lack both proteins throughout the brain and from the earliest stages of development. Indeed, more recent work supports conclusions from the ΔFosB overexpressing mice: inducible overexpression of a truncated mutant of c-Jun, which acts as a dominant negative antagonist of ΔFosB, selectively in nucleus accumbens and dorsal striatum shows reduced sensitivity to the rewarding effects of cocaine.¶ These findings emphasize the caution that must be used in interpreting results from mice with constitutive mutations and illustrate the importance of mice with inducible and cell type-specific mutations in studies of plasticity in the adult brain.
Target Genes for ΔFosB
Because ΔFosB is a transcription factor, presumably the protein causes behavioral plasticity through alterations in the expression of other genes. ΔFosB is generated by alternative splicing of the fosB gene and lacks a portion of the C-terminal transactivation domain present in full-length FosB. As a result, it was originally proposed that ΔFosB functions as a transcriptional repressor (29). However, work in cell culture has demonstrated clearly that ΔFosB can either induce or repress AP-1-mediated transcription depending on the particular AP-1 site used (21, 29–31). Full-length FosB exerts the same effects as ΔFosB on certain promoter fragments, but different effects on others. Further work is needed to understand the mechanisms underlying these varied actions of ΔFosB and FosB.
Our group has used two approaches to identify target genes for ΔFosB. One is the candidate gene approach. We initially considered α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors as putative targets, given the important role of glutamatergic transmission in the nucleus accumbens. Work to date has indicated that one particular AMPA glutamate receptor subunit, GluR2, may be a bona fide target for ΔFosB (Fig. 2). GluR2 expression, but not the expression of other AMPA receptor subunits, is increased in nucleus accumbens (but not dorsal striatum) upon overexpression of ΔFosB (28), and expression of a dominant negative mutant attenuates the ability of cocaine to induce the protein.¶ In addition, the promoter of the GluR2 gene contains a consensus AP-1 site that binds ΔFosB (28). Overexpression of GluR2 in the nucleus accumbens, by use of viral-mediated gene transfer, increases an animal’s sensitivity to the rewarding effects of cocaine, thereby mimicking part of the phenotype seen in the ΔFosB-expressing mice (28). Induction of GluR2 could account for the reduced electrophysiological sensitivity of nucleus accumbens neurons to AMPA receptor agonists after chronic cocaine administration (32), because AMPA receptors containing GluR2 show reduced overall conductance and reduced Ca2+ permeability. Reduced responsiveness of these neurons to excitatory inputs may then enhance responses to a drug of abuse. However, the ways in which dopaminergic and glutamatergic signals in nucleus accumbens regulate addictive behavior remain unknown; this will require a neural circuit level of understanding, which is not yet available.
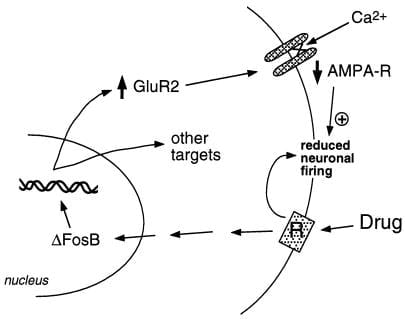
Figure 2
The AMPA glutamate receptor subunit, GluR2, is a putative target for ΔFosB. Shown is how ΔFosB-mediated induction of GluR2 may alter the physiological responsiveness of nucleus accumbens neurons and lead to sensitized responses to drugs of abuse. According to this scheme, drugs of abuse produce their acute reinforcing effects via inhibition of nucleus accumbens neurons. With repeated exposure, the drugs induce ΔFosB, which regulates numerous target genes, including GluR2. This increases the proportion of AMPA receptors (AMPA-R) on nucleus accumbens neurons that contain the GluR2 subunit, which causes reduced overall AMPA current and reduced Ca2+ current. This reduced excitability could render the neurons more sensitive to the acute inhibitory effects of the drugs and thereby to the drugs’ reinforcing effects.
Another putative target for ΔFosB is the gene encoding dynorphin. As stated earlier, dynorphin is expressed in the subset of nucleus accumbens medium spiny neurons that show induction of ΔFosB. Dynorphin appears to function in an intercellular feedback loop: its release inhibits the dopaminergic neurons that innervate the medium spiny neurons, via κ opioid receptors present on dopaminergic nerve terminals in the nucleus accumbens and also on cell bodies and dendrites in the ventral tegmental area (Fig. 3) (33–35). This idea is consistent with the ability of a κ receptor agonist, upon administration into either of these two brain regions, to decrease drug reward (35).
Recent work has indicated that ΔFosB decreases the expression of dynorphin,‖ which could contribute to the enhancement of reward mechanisms seen with ΔFosB induction. Interestingly, another drug-regulated transcription factor, CREB (cAMP response element binding protein) (2, 3), exerts the opposite effect: it induces dynorphin expression in the nucleus accumbens and reduces the rewarding properties of cocaine and morphine (4).**
Because drug-induced activation of CREB dissipates rapidly after drug administration, such reciprocal regulation of dynorphin by CREB and ΔFosB could explain the reciprocal behavioral changes that occur during early and late phases of withdrawal, with negative emotional symptoms and reduced drug sensitivity predominating during early phases of withdrawal, and sensitization to the rewarding and incentive motivational effects of drugs predominating at later time points.
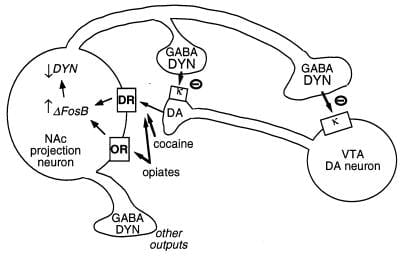
Figure 3
Dynorphin is a putative target for ΔFosB. Shown is a ventral tegmental area (VTA) dopamine (DA) neuron innervating a class of nucleus accumbens (NAc) GABAergic projection neuron that expresses dynorphin (DYN). Dynorphin serves a feedback mechanism in this circuit: dynorphin, released from terminals of the NAc neurons, acts on κ opioid receptors located on nerve terminals and cell bodies of the DA neurons to inhibit their functioning. ΔFosB, by inhibiting dynorphin expression, may down-regulate this feedback loop and enhance the rewarding properties of drugs of abuse. Not shown is the reciprocal effect of CREB on this system: CREB enhances dynorphin expression and thereby attenuates the rewarding properties of drugs of abuse (4). GABA, γ-aminobutyric acid; DR, dopamine receptor; OR, opioid receptor.
The second approach used to identify target genes for ΔFosB involves DNA microarray analysis. Inducible overexpression of ΔFosB increases or decreases the expression of numerous genes in the nucleus accumbens (36). Although considerable work is now needed to validate each of these genes as physiologic targets of ΔFosB and to understand their contribution to the addiction phenotype, one important target appears to be Cdk5 (cyclin-dependent kinase-5). Thus, Cdk5 was initially identified as ΔFosB-regulated by use of microarrays, and later shown to be induced in nucleus accumbens and dorsal striatum after chronic cocaine administration (37). ΔFosB activates the cdk5 gene via an AP-1 site present within the gene’s promoter (36). Together, these data support a scheme wherein cocaine induces Cdk5 expression in these brain regions via ΔFosB. Induction of Cdk5 appears to alter dopaminergic signaling at least in part via increased phosphorylation of DARPP-32 (37), which is converted from an inhibitor of protein phosphatase-1 to an inhibitor of protein kinase A upon its phosphorylation by Cdk5 (26).
Role of ΔFosB in Mediating “Permanent” Plasticity to Drugs of Abuse
Although the ΔFosB signal is relatively long-lived, it is not permanent. ΔFosB degrades gradually and can no longer be detected in brain after 1–2 months of drug withdrawal, even though certain behavioral abnormalities persist for much longer periods of time. Therefore, ΔFosB per se would not appear to be able to mediate these semipermanent behavioral abnormalities. The difficulty in finding the molecular adaptations that underlie the extremely stable behavioral changes associated with addiction is analogous to the challenges faced in the learning and memory field. Although there are elegant cellular and molecular models of learning and memory, it has not to date been possible to identify molecular and cellular adaptations that are sufficiently long-lived to account for highly stable behavioral memories. Indeed, ΔFosB is the longest-lived adaptation known to occur in adult brain, not only in response to drugs of abuse, but to any other perturbation (that doesn’t involve lesions) as well. Two proposals have evolved, both in the addiction and learning and memory fields, to account for this discrepancy.
One possibility is that more transient changes in gene expression, such as those mediated via ΔFosB or other transcription factors (e.g., CREB), may mediate more long-lived changes in neuronal morphology and synaptic structure. For example, an increase in the density of dendritic spines (particularly an increase in two-headed spines) accompanies the increased efficacy of glutamatergic synapses at hippocampal pyramidal neurons during long-term potentiation (38–40), and parallels the enhanced behavioral sensitivity to cocaine mediated at the level of medium spiny neurons of the nucleus accumbens (41). It is not known whether such structural changes are sufficiently long-lived to account for highly stable changes in behavior, although the latter persist for at least 1 month of drug withdrawal. Recent evidence raises the possibility that ΔFosB, and its induction of Cdk5, is one mediator of drug-induced changes in synaptic structure in the nucleus accumbens (Fig. 4).‡‡ Thus, infusion of a Cdk5 inhibitor into the nucleus accumbens prevents the ability of repeated cocaine exposure to increase dendritic spine density in this region. This is consistent with the view that Cdk5, which is enriched in brain, regulates neural structure and growth (see refs. 36 and 37). It is possible, although by no means proven, that such changes in neuronal morphology may outlast the ΔFosB signal itself.
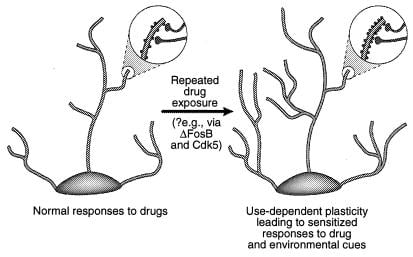
Figure 4
Regulation of dendritic structure by drugs of abuse. Shown is the expansion of a neuron’s dendritic tree after chronic exposure to a drug of abuse, as has been observed with cocaine in the nucleus accumbens and prefrontal cortex (41). The areas of magnification show an increase in dendritic spines, which is postulated to occur in conjunction with activated nerve terminals. This increase in dendritic spine density may be mediated via ΔFosB and the consequent induction of Cdk5 (see text). Such alterations in dendritic structure, which are similar to those observed in some learning models (e.g., long-term potentiation), could mediate long-lived sensitized responses to drugs of abuse or environmental cues. [Reproduced with permission from ref. 3 (Copyright 2001, Macmillian Magazines Ltd.)].
Another possibility is that the transient induction of a transcription factor (e.g., ΔFosB, CREB) leads to more permanent changes in gene expression through the modification of chromatin. These and many other transcription factors are believed to activate or repress the transcription of a target gene by promoting the acetylation or deacetylation, respectively, of histones in the vicinity of the gene (42). Although such acetylation and deacetylation of histones can apparently occur very rapidly, it is possible that ΔFosB or CREB might produce longer-lasting adaptations in the enzymatic machinery that controls histone acetylation. ΔFosB or CREB may also promote longer-lived changes in gene expression by regulating other modifications of chromatin (e.g., DNA or histone methylation) that have been implicated in the permanent changes in gene transcription that occur during development (see refs. 42 and 43). Although these possibilities remain speculative, they could provide a mechanism by which transient adaptations to a drug of abuse (or some other perturbation) lead to essentially life-long behavioral consequences.
References
- ↵
- Nestler E J,
- Hope B T,
- Widnell K L
(1993) Neuron 11:995–1006.
- ↵
- Berke J D,
- Hyman S E
(2000) Neuron 25:515–532.
- ↵
- Nestler E J
(2001) Nat Rev Neurosci 2:119–128.
- ↵
- Carlezon W A Jr,
- Thome J,
- Olson V G,
- Lane-Ladd S B,
- Brodkin E S,
- Hiroi N,
- Duman R S,
- Neve R L,
- Nestler E J
(1998) Science 282:2272–2275.
-
- O’Donovan K J,
- Tourtellotte W G,
- Millbrandt J,
- Baraban J M
(1999) Trends Neurosci 22:167–173.
- ↵
- Mackler S A,
- Korutla L,
- Cha X Y,
- Koebbe M J,
- Fournier K M,
- Bowers M S,
- Kalivas P W
(2000) J Neurosci 20:6210–6217.
- ↵
- Morgan J I,
- Curran T
(1995) Trends Neurosci 18:66–67.
- ↵
- Young S T,
- Porrino L J,
- Iadarola M J
(1991) Proc Natl Acad Sci USA 88:1291–1295.
-
- Graybiel A M,
- Moratalla R,
- Robertson H A
(1990) Proc Natl Acad Sci USA 87:6912–6916.
-
- Hope B,
- Kosofsky B,
- Hyman S E,
- Nestler E J
(1992) Proc Natl Acad Sci USA 89:5764–5768.
- ↵
- Kelz M B,
- Nestler E J
(2000) Curr Opin Neurol 13:715–720.
- ↵
- Koob G F,
- Sanna P P,
- Bloom F E
(1998) Neuron 21:467–476.
- ↵
- Wise R A
(1998) Drug Alcohol Dependence 51:13–22.
- ↵
- Hope B T,
- Nye H E,
- Kelz M B,
- Self D W,
- Iadarola M J,
- Nakabeppu Y,
- Duman R S,
- Nestler E J
(1994) Neuron 13:1235–1244.
- ↵
- Nye H,
- Hope B T,
- Kelz M,
- Iadarola M,
- Nestler E J
(1995) J Pharmacol Exp Ther 275:1671–1680.
-
- Nye H E,
- Nestler E J
(1996) Mol Pharmacol 49:636–645.
- ↵
- Moratalla R,
- Elibol B,
- Vallejo M,
- Graybiel A M
(1996) Neuron 17:147–156.
- ↵
- Pich E M,
- Pagliusi S R,
- Tessari M,
- Talabot-Ayer D,
- Hooft van Huijsduijnen R,
- Chiamulera C
(1997) Science 275:83–86.
- ↵
- Chen J S,
- Nye H E,
- Kelz M B,
- Hiroi N,
- Nakabeppu Y,
- Hope B T,
- Nestler E J
(1995) Mol Pharmacol 48:880–889.
- ↵
- Hiroi N,
- Brown J,
- Ye H,
- Saudou F,
- Vaidya V A,
- Duman R S,
- Greenberg M E,
- Nestler E J
(1998) J Neurosci 18:6952–6962.
- ↵
- Chen J,
- Kelz M B,
- Hope B T,
- Nakabeppu Y,
- Nestler E J
(1997) J Neurosci 17:4933–4941.
- ↵
- Hiroi N,
- Brown J,
- Haile C,
- Ye H,
- Greenberg M E,
- Nestler E J
(1997) Proc Natl Acad Sci USA 94:10397–10402.
- ↵
- Fienberg A A,
- Hiroi N,
- Mermelstein P,
- Song W-J,
- Snyder G L,
- Nishi A,
- Cheramy A,
- O’Callaghan J P,
- Miller D,
- Cole D G,
- et al.
(1998) Science 281:838–842.
- ↵
- Hiroi N,
- Feinberg A,
- Haile C,
- Greengard P,
- Nestler E J
(1999) Eur J Neurosci 11:1114–1118.
- ↵
- Greengard P,
- Allen P B,
- Nairn A C
(1999) Neuron 23:435–447.
- ↵
- Bibb J A,
- Snyder G L,
- Nishi A,
- Yan Z,
- Meijer L,
- Fienberg A A,
- Tsai L H,
- Kwon Y T,
- Girault J A,
- Czernik A J,
- et al.
(1999) Nature (London) 402:669–671.
- ↵
- Chen J S,
- Kelz M B,
- Zeng G Q,
- Sakai N,
- Steffen C,
- Shockett P E,
- Picciotto M,
- Duman R S,
- Nestler E J
(1998) Mol Pharmacol 54:495–503.
- ↵
- Kelz M B,
- Chen J S,
- Carlezon W A,
- Whisler K,
- Gilden L,
- Beckmann A M,
- Steffen C,
- Zhang Y-J,
- Marotti L,
- Self S W,
- et al.
(1999) Nature (London) 401:272–276.
- ↵
- Dobrazanski P,
- Noguchi T,
- Kovary K,
- Rizzo C A,
- Lazo P S,
- Bravo R
(1991) Mol Cell Biol 11:5470–5478.
-
- Nakabeppu Y,
- Nathans D
(1991) Cell 64:751–759.
- ↵
- Yen J,
- Wisdom R M,
- Tratner I,
- Verma I M
(1991) Proc Natl Acad Sci USA 88:5077–5081.
- ↵
- White F J,
- Hu X-T,
- Zhang X-F,
- Wolf M E
(1995) J Pharmacol Exp Ther 273:445–454.
- ↵
- Hyman S E
(1996) Neuron 16:901–904.
-
- Kreek M J
(1997) Pharmacol Biochem Behav 57:551–569.
- ↵
- Shippenberg T S,
- Rea W
(1997) Pharmacol Biochem Behav 57:449–455.
- ↵
- Chen J S,
- Zhang Y J,
- Kelz M B,
- Steffen C,
- Ang E S,
- Zeng L,
- Nestler E J
(2000) J Neurosci 20:8965–8971.
- ↵
- Bibb J A,
- Chen J S,
- Taylor J R,
- Svenningsson P,
- Nishi A,
- Snyder G L,
- Yan Z,
- Sagawa Z K,
- Nairn A C,
- Nestler E J,
- et al.
(2001) Nature (London) 410:376–380.
- ↵
- Luscher C,
- Nicoll R A,
- Malenka R C,
- Muller D
(2000) Nat Neurosci 3:545–550.
-
- Malinow R,
- Mainen Z F,
- Hayashi Y
(2000) Curr Opin Neurobiol 10:352–357.
- ↵
- Scannevin R H,
- Huganir R L
(2000) Nat Rev Neurosci 1:133–141.
Robinson, T. E. & Kolb, B. (1999) (1997) Eur. J. Neurosci.11, 1598–1604.
- ↵
- Carey M,
- Smale S T
(2000) Transcriptional Regulation in Eukaryotes (Cold Spring Harbor Lab. Press, Plainview, NY).
- ↵
- Spencer V A,
- Davie J R
(1999) Gene 240:1–12.
 Facebook
Facebook Twitter
Twitter- Google+
 CiteULike
CiteULike Delicious
Delicious Digg
Digg Mendeley
Mendeley
HighWire Press-hosted articles citing this article
- Natural and Drug Rewards Act on Common Neural Plasticity Mechanisms with {Delta}FosB as a Key Mediator J. Neurosci. 2013 33 (8) 3434-3442
- Drugs, Crime, and the Epigenetics of Hedonic Allostasis Journal of Contemporary Criminal Justice 2012 28 (3) 314-328
- Abstract
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Abstract
- Full Text (HTML)
- Full Text (PDF)
- Morphine Activates the E Twenty Six-Like Transcription Factor-1/Serum Response Factor Pathway via Extracellular Signal-Regulated Kinases 1/2 in F11 Cells Derived from Dorsal Root Ganglia Neurons J. Pharmacol. Exp. Ther. 2012 342 (1) 41-52
- Molecular Mechanism for a Gateway Drug: Epigenetic Changes Initiated by Nicotine Prime Gene Expression by Cocaine Sci Transl Med 2011 3 (107) 107ra109
- Enhanced Sucrose and Cocaine Self-Administration and Cue-Induced Drug Seeking after Loss of VGLUT2 in Midbrain Dopamine Neurons in Mice J. Neurosci. 2011 31 (35) 12593-12603
- Chronic intermittent hypoxia increases blood pressure and expression of FosB/{Delta}FosB in central autonomic regions Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011 301 (1) R131-R139
- Absence of the GPR37/PAEL receptor impairs striatal Akt and ERK2 phosphorylation, {Delta}FosB expression, and conditioned place preference to amphetamine and cocaine FASEB J. 2011 25 (6) 2071-2081
- The Relationship between Duration of Initial Alcohol Exposure and Persistence of Molecular Tolerance Is Markedly Nonlinear J. Neurosci. 2011 31 (7) 2436-2446
- In Vivo Bioluminescence Imaging Reveals Redox-Regulated Activator Protein-1 Activation in Paraventricular Nucleus of Mice With Renovascular Hypertension Hypertension 2011 57 (2) 289-297
- Striatal Overexpression of {Delta}FosB Reproduces Chronic Levodopa-Induced Involuntary Movements J. Neurosci. 2010 30 (21) 7335-7343
- Epigenetic Mediation of Environmental Influences in Major Psychotic Disorders Schizophr Bull 2009 35 (6) 1045-1056
- DNA-Based MRI Probes for Specific Detection of Chronic Exposure to Amphetamine in Living Brains J. Neurosci. 2009 29 (34) 10663-10670
- Altered Dendritic Spine Plasticity in Cocaine-Withdrawn Rats J. Neurosci. 2009 29 (9) 2876-2884
- Overexpression Screen in Drosophila Identifies Neuronal Roles of GSK-3{beta}/shaggy as a Regulator of AP-1-Dependent Developmental Plasticity Genetics 2008 180 (4) 2057-2071
- Transcription MRI: A New View of the Living Brain Neuroscientist 2008 14 (5) 503-520
- {Delta}FosB Induction in Orbitofrontal Cortex Mediates Tolerance to Cocaine-Induced Cognitive Dysfunction J. Neurosci. 2007 27 (39) 10497-10507
- Enduring vulnerability to reinstatement of methamphetamine-seeking behavior in glial cell line-derived neurotrophic factor mutant mice FASEB J. 2007 21 (9) 1994-2004
- {Delta}FosB in the Nucleus Accumbens Regulates Food-Reinforced Instrumental Behavior and Motivation J. Neurosci. 2006 26 (36) 9196-9204
- Regulation of {Delta}FosB Stability by Phosphorylation. J. Neurosci. 2006 26 (19) 5131-5142
- Expression of Mutant NMDA Receptors in Dopamine D1 Receptor-Containing Cells Prevents Cocaine Sensitization and Decreases Cocaine Preference J. Neurosci. 2005 25 (28) 6651-6657
- D1 Dopamine Receptors Modulate {Delta}FosB Induction in Rat Striatum after Intermittent Morphine Administration J. Pharmacol. Exp. Ther. 2005 314 (1) 148-154
- Neurobiology of Mice Selected for High Voluntary Wheel-running Activity Integr. Comp. Biol. 2005 45 (3) 438-455
- Effects of water deprivation and rehydration on c-Fos and FosB staining in the rat supraoptic nucleus and lamina terminalis region Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005 288 (1) R311-R321
- Transcriptional Induction of FosB/{Delta}FosB Gene by Mechanical Stress in Osteoblasts J Biol Chem 2004 279 (48) 49795-49803
- Induction of {Delta}FosB in Reward-Related Brain Structures after Chronic Stress J. Neurosci. 2004 24 (47) 10594-10602
- Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice Am. J. Physiol. Endocrinol. Metab. 2004 287 (1) E105-E113
- DNA microarray analysis of gene expression in human optic nerve head astrocytes in response to hydrostatic pressure Physiol. Genomics 2004 17 (2) 157-169
- Superoxide Is Involved in the Central Nervous System Activation and Sympathoexcitation of Myocardial Infarction-Induced Heart Failure Circ. Res. 2004 94 (3) 402-409
- Adenosine A2A receptors in neuroadaptation to repeated dopaminergic stimulation: Implications for the treatment of dyskinesias in Parkinson’s disease Neurology 2003 61 (90116) S74-81
- Cytoplasmic Versus Nuclear Localization of Fos-Related Proteins in the Frog, Rana esculenta, Testis: In Vivo and Direct In Vitro Effect of a Gonadotropin-Releasing Hormone Agonist Biol. Reprod. 2003 68 (3) 954-960
- Periadolescent Mice Show Enhanced Delta FosB Upregulation in Response to Cocaine and Amphetamine J. Neurosci. 2002 22 (21) 9155-9159
- Delta FosB Regulates Wheel Running J. Neurosci. 2002 22 (18) 8133-8138
- CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli Proc. Natl. Acad. Sci. USA 2002 99 (17) 11435-11440
- Psychogenomics: Opportunities for Understanding Addiction J. Neurosci. 2001 21 (21) 8324-8327