Comments: This review was produced by the head of NIDA, Nora Volkow, and her team. It leaves little doubt that chemical addictions and behavioral addictions share the same or similar mechanisms and neural circuitry. This makes perfect sense since chemical addictions hijack the neural circuitry for bonding, sex, and eating. Since sex releases twice as much dopamine as eating your favorite food, and a porn user can keep dopamine elevated for hours, it crazy to propose that porn addiction cannot exist.
Curr Top Behav Neurosci. 2011 Oct 21.
Volkow ND, Wang GJ, Fowler JS, Tomasi D, Baler R.
Source
National Institute on Drug Abuse, 6001 Executive Boulevard 6001, Room 5274, Bethesda, MD, 20892, USA, [email protected].
Abstract
Both drug addiction and obesity can be defined as disorders in which the saliency value of one type of reward (drugs and food, respectively) becomes abnormally enhanced relative to, and at the expense of others. This model is consistent with the fact that both drugs and food have powerful reinforcing effects-partly mediated by dopamine increases in the limbic system-that, under certain circumstances or in vulnerable individuals, could overwhelm the brain’s homeostatic control mechanisms. Such parallels have generated significant interest in understanding the shared vulnerabilities and trajectories between addiction and obesity. Now, brain imaging discoveries have started to uncover common features between these two conditions and to delineate some of the overlapping brain circuits whose dysfunctions may explain stereotypic and related behavioral deficits in human subjects. These results suggest that both obese and drug-addicted individuals suffer from impairments in dopaminergic pathways that regulate neuronal systems associated not only with reward sensitivity and incentive motivation, but also with conditioning (memory/learning), impulse control (behavioural inhibition), stress reactivity, and interoceptive awareness. Here, we integrate findings predominantly derived from positron emission tomography that shed light on the role of dopamine in drug addiction and in obesity, and propose an updated working model to help identify treatment strategies that may benefit both of these conditions.
1 Background
2 The Role of Dopamine in Acute Reward to Drugs and Food
3 Imaging DA in Response to Drugs and to Conditioned Cues in Addiction
4 The Impact of Dysfunction in Inhibitory Control
5 Involvement of Motivation Circuits
6 Involvement of Interoceptive Circuitry
7 The Circuitry of Aversion
8 Pathological Drug and Food Reward: An Updated Working Model
1 Background
Dopamine (DA) is considered a key to the rewarding effects of natural and drug rewards. However, its role in the loss of control and compulsive behaviours that are associated with addiction and obesity are much less clear. PET studies have played a crucial role in characterizing the role of the brain DA systems in addiction (in addition to its role in drug reward) and in obesity. Indeed, drugs of abuse (including alcohol) are consumed by humans or self-administered by laboratory animals because they are inherently rewarding, an effect that is mediated through their DA-enhancing properties in the mesolimbic system (Wise 2009). However, in the case of addiction, imaging studies have revealed that the disorder affects not only the DA reward circuit but also other DA pathways involved in the modulation of conditioning/habits, motivation, and executive functions (inhibitory control, salience attribution, and decision-making), and that DA deficits may also participate in the enhanced stress reactivity and disruption of interoceptive awareness associated with addiction. Preclinical and clinical studies have also revealed other neurotransmitters (and neuropeptides) that play important roles in drug reward and addiction (i.e., cannabinoids, opioids) and are intimately involved in the neuroplastic changes that follow repeated drug use (i.e., glutamate, opioids, GABA, corticotropin-releasing factor). The glutamatergic system is particularly prominent in this regard because it mediates the disruptions in both long-term potentiation and long-term depression that have been observed in animal models of chronic drug administration (Thomas et al. 2008). Reviews pertaining to these additional systems can be found elsewhere (Kalivas 2009; Koob 1992).
Since drugs activate the same reward systems that underlie food reward, it is not totally unexpected that, in general, brain imaging studies have supported the notion that impairments in DA-modulated circuits be also implicated in pathologic, compulsive eating behaviours. Food cues, like drug cues, increase striatal extracellular DA and drive the motivation to engage in the behaviours that are necessary to procure and eat the food, providing evidence for the involvement of DA not just in food reward but also in the non-hedonic motivational properties of food (i.e., caloric requirements) and the decrease in inhibitory control seen in compulsive overeating (Avena et al. 2008; Volkow et al. 2008a).
Here, we review findings from imaging studies that specifically focus on the overlaps in the brain circuits that are disrupted in obesity and in drug addiction. It is worth remembering, however, that the regulation of food intake behaviours is much more complex than the regulation of drug intake. The latter is predominantly mediated by the rewarding effects of drugs whereas the former is modulated not just by its rewarding effects (hedonic factors) but also by multiple peripheral and central factors that sense nutrient requirements in the body necessary for survival (homeostatic factors). Interestingly, there is growing evidence that homeostatic factors (e.g., insulin, leptin, ghrelin) modulate food intake in part by increasing or decreasing the sensitivity of brain reward circuits to food stimuli (Volkow et al. 2011a).
2 The Role of Dopamine in Acute Reward to Drugs and Food
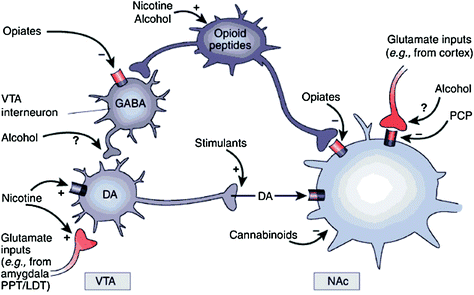
Whether directly or indirectly, all addictive drugs display an ability to increase DA in nucleus accumbens (NAc) via specific interactions with different molecular targets (Nestler 2004) (Fig. 1). The mesolimbic DA pathway [DA cells in ventral tegmental area (VTA) that project into the NAc] seems to be crucial for drug reward (Wise 2009). However, as described below, other DA pathways [mesostriatal (DA cells in substantia nigra projecting into dorsal striatum) and mesocortical (DA cells in VTA projecting into frontal cortex)] also contribute to drug reward and addiction (Wise 2009). Overall, it appears that the rewarding and conditioning effects of drugs are predominantly driven by phasic DA cell firing, which leads to large and transient DA increases. In contrast, the downstream changes in executive function that occur in addiction are linked with changes in tonic DA cell firing and result in lower but more stable DA levels (Grace 2000; Wanat et al. 2009). This, in turn, point to the D1 receptors (D1R), which are low affinity DA receptors that stimulate cyclic AMP signaling, as being involved both in acute drug reward as well as in conditioning, since these are associated with the high DA concentrations necessary to stimulate D1R. In contrast, D2Rs, which inhibit cyclic AMP signaling, are stimulated by both phasic and tonic DA. Note that, due to the lack of specific radiotracers for the PET imaging of DA receptors of the D1, D3, D4, and D5 types, most studies on the effects of drugs of abuse and addiction in the human brain have focused on D2Rs.
Fig. 1 Drugs of abuse act on the reward and ancillary circuits through different mechanisms, however, they all lead to similar dopaminergic effects in the VTA and NAc. Thus, stimulants boost acumbal DA directly, while opiates do this by lowering the inhibitory tone of GABAergic interneurons on DA signaling both either in the VTA or in then NAc. While the mechanisms of other drugs of abuse is less clear, there is evidence suggesting that nicotine may activate VTA DA directly through nicotinic acetylcholine receptor (nAChR) on those neurons and indirectly via stimulation of its receptors on glutamatergic nerve terminals that innervate the DA cells. Alcohol appears to inhibit GABAergic terminals in VTA, leading to DA neurons disinhibition in the VTA. Cannabinoids act, among others, through the activation of CB1 receptors on glutamatergic and GABAergic nerve terminals in the NAc, and on NAc neurons themselves. Phencyclidine (PCP) may act by inhibiting postsynaptic NMDA glutamate receptors in the NAc. In addition, there is some evidence suggesting that nicotine and alcohol may also interact with endogenous opioid and cannabinoid pathways (not shown). PPT/LDT, peduncular pontine tegmentum/lateral dorsal tegmentum. Reprinted with permission Nestler (2005)
In humans, PET studies have shown that several drugs [stimulants (Drevets et al. 2001; Volkow et al. 1999b), nicotine (Brody et al. 2009), alcohol (Boileau et al. 2003), and marijuana (Bossong et al. 2009)] increase DA in dorsal and ventral striatum (where the NAc is located). These studies take advantage of several radiotracers, such as [11C]raclopride, that bind to D2R but only when these are not binding endogenous DA (unoccupied), which under baseline conditions corresponds to 85–90% of the striatal D2R (Abi-Dargham et al. 1998). Thus, a comparison of [11C]raclopride binding after placebo and after drug administration can help us estimate the decreases in D2R availability induced by the drug (or other stimuli that can increase DA). These decreases in [11C]raclopride binding are proportional to the DA increases (Breier et al. 1997). These studies have shown that drug-induced DA increases in striatum are proportional to the intensity of the subjective experience of euphoria or “high” [see review (Volkow et al. 2009a)] (Fig. 2).
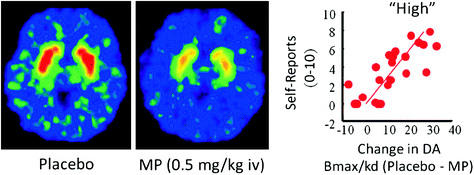
Fig. 2 Effects of intravenous methylphenidate (MP) in raclopride binding and relationship between striatal DA increases induced by MP in the striatum and the self-reports of “high”. Modified from Volkow et al. (1999b)
PET studies have also revealed a clear, direct relationship between a drug’s pharmacokinetic profile (i.e., the speed with which it enters and leaves the brain) and its reinforcing effects. Specifically, the faster a drug reaches peak levels in the brain the more intense the “high” (Volkow et al. 2009a). For example, for an equivalent level of cocaine reaching the brain (assessed through PET), when cocaine entered the brain rapidly (smoked or i.v. administration), it elicited a more intense “high” than when it entered at a slower rate (snorted) (Volkow et al. 2000). This is consistent with preclinical studies showing a similar correlation between a drug’s pharmacokinetic profile and its reinforcing properties (Balster and Schuster 1973). It is reasonable to hypothesize that such abrupt and large DA increases as triggered by drugs of abuse may mimic the fast and large DA increases that result from phasic DA firing that have been associated, in the brain, with the processing of information about reward and saliency (Schultz 2010). Such drug-induced DA increases in the NAc may be necessary for addiction, but the fact that they occur also in non-addicted individuals indicates that they are insufficient to explain the impulsive and compulsive drug use characteristic of addiction.
There is now evidence that comparable dopaminergic responses are linked with food reward and that these mechanisms are also likely to play a role in excessive food consumption and obesity. It is well known that certain foods, particularly those rich in sugars and fat, are potently rewarding (Lenoir et al. 2007). High-calorie foods can promote over-eating (eating that is uncoupled from energetic needs) and trigger learned associations between the stimulus and the reward (conditioning). In evolutionary terms, this property of palatable foods used to be advantageous in environments where food sources were scarce and/or unreliable, because it ensured that food was eaten when available, enabling energy to be stored in the body (as fat) for future use. Unfortunately, in societies like ours, where food is plentiful and constantly available, this adaptation has become a liability.
Several neurotransmitters, including DA, cannabinoids, opioids, and serotonin, as well as hormones and neuropeptides involved in homeostatic regulation of food intake, such as insulin, orexin, leptin, and ghrelin, have been implicated in the rewarding effects of food (Atkinson 2008; Cason et al. 2010; Cota et al. 2006). Of these, DA has been the most thoroughly investigated and is the best characterized. Experiments in rodents have shown that, upon first exposure to a food reward, the firing of DA neurons in the VTA increases with a resulting increase in DA release in NAc (Norgren et al. 2006). Similarly, in healthy, normal-weight human subjects, the ingestion of palatable food has been shown to release DA in the dorsal striatum in proportion to the ratings of meal pleasantness (Small et al. 2003) (Fig. 3). However, and as seen in studies with drug abusers, food-induced increases in striatal DA alone cannot explain the difference between normal food intake and excessive compulsive food consumption since these also occur in healthy individuals who do not eat excessively. Thus, as is the case for addiction, downstream adaptations are likely to be involved in the loss of control over food intake.
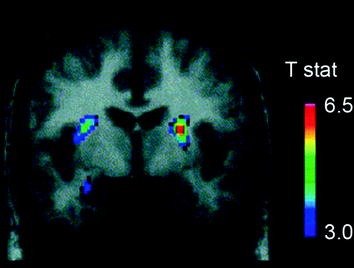
Fig. 3 Dopamine release induced by feeding. Coronal section from the T-map of statistically significant reductions in [11C]raclopride’s binding potential (BP) following feeding. The colour bar represents the t statistic values. (Reprinted with permission Small et al. 2003)
3 Imaging DA in Response to Drugs and to Conditioned Cues in Addiction
DA’s role in reinforcement is more complex than just coding for reward per se (hedonic pleasure); for example, stimuli that induce fast and large DA increases also trigger conditioned responses and elicit incentive motivation to procure them (Owesson-White et al. 2009). This is important because, through the process of conditioning, neutral stimuli that are linked to the reinforcer (whether a natural or a drug reinforcer) acquire the ability by themselves to increase DA in striatum (including NAc) in anticipation of the reward, thus engendering a strong motivation to seek the drug (Owesson-White et al. 2009). However, uncoupling reward and conditioning mechanisms in the process of drug addiction is more challenging than for food consumption because drugs of abuse, through their pharmacological effects, directly activate DA neurons (i.e., nicotine) or increase DA release (i.e., amphetamine).
Brain imaging studies that compared the DA increases induced by the stimulant drug methylphenidate (MP) or amphetamine (AMPH) among cocaine addicted subjects vs. controls showed a marked attenuation of MP or AMPH-induced DA increases in striatum (50% lower in detoxified abusers and 80% in active abusers) and lower self-reports of the drug’s rewarding effects relative to non-drug-abusing controls (Martinez et al. 2007; Volkow et al. 1997) (Fig. 4). This was surprising since MP and AMPH are pharmacologically similar to cocaine and methamphetamine, respectively, and drug abusers cannot distinguish between them when they are administered intravenously. Since the marked reductions in the drug-induced DA increases were observed whether the cocaine abusers had been detoxified or not, this indicates that the state of withdrawal is not a confounding factor (Volkow et al. 2011b). These and related results (Volkow et al. 2009a) are consistent with the hypothesis that the hedonic response becomes deficient in drug-addicted individuals, and further strengthen the notion that the acute pharmacological DA-enhancing effects of the drug in NAc cannot explain by themselves the increased motivation to consume them.
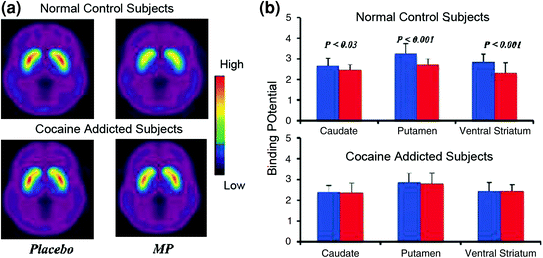
Fig. 4 DA changes induced by i.v. MP in controls and in active cocaine-addicted subjects. a Average nondisplaceable biding potential (BPND) images of [11C]raclopride in active cocaine-addicted subjects (n = 19) and in controls (n = 24) tested after placebo and after i.v. MP. b D2R availability (BPND) in caudate, putamen, and ventral striatum after placebo (blue) and after MP (red) in controls and in cocaine-addicted subjects. MP reduced D2R in controls but not in cocaine-addicted subjects. Note that cocaine abusers show both decreases in baseline striatal D2R availability (placebo measure) and decreases in DA release when given i.v. MP (measured as decreases in D2R availability from baseline). Although one could question the extent to which the low striatal D2R availability in cocaine-addicted subject limits the ability to detect further decreases from MP, the fact that cocaine-addicted subjects show reductions in D2R availability when exposed to cocaine cues indicates that the attenuated effects of MP on [11C]raclopride binding reflect decreased DA release. Reprinted with permission (Volkow et al. 1997; Wang et al. 2010)
The response of VTA DA neurons to rewarding stimuli changes with repeated exposure.
While DA cells fire upon the first exposure to a novel reward, repeated exposure to DA causes the neurons to stop firing upon reward consumption and fire instead when they are exposed to stimuli that are predictive of the reward (Schultz et al. 1997). This is likely to underlie DA’s role in learning and conditioning. Indeed, drug-induced phasic DA signaling can eventually trigger neuroadaptations in ancillary circuits that are related to habit formation and behavioural conditioning. These changes are predominantly induced by D1R signaling and synaptic changes in glutamate-modulated NMDA and AMPA receptors (Luscher and Malenka 2011; Zweifel et al. 2009). Recruitment of these circuits is significant in disease progression because the ensuing conditioned responses help explain the intense desire for the drug (craving) and the compulsive use that occurs when addicted subjects are exposed to drug-associated cues. This hypothesis is consistent with independent observations (Volkow et al. 2006b; Wong et al. 2006) that show the power of cocaine-associated cue exposure to raise DA levels in the dorsal striatum and trigger a concomitant increase in the subjective experience of craving in detoxified cocaine abusers (Fig. 5). Since the dorsal striatum plays a role in habit learning (Belin et al. 2009; Yin et al. 2004), the association is likely to reflect the strengthening of habits as chronicity of addiction progresses. This suggests that a basic disruption in addiction might relate to the DA-triggered conditioned responses that result in habits leading to intense craving and compulsive drug consumption. Interestingly, in actively using cocaine-addicted subjects, the DA increases triggered by conditioned cues appear to be even larger than those produced by the stimulant drug itself as assessed in two separate group of subjects (Volkow et al. 2011b, 2006b), suggesting that conditioned responses may drive the DA signaling that maintains the motivation to take the drug even when its pharmacological effects appear to be reduced. Thus, although drugs may initially induce feelings of immediate reward through DA release in the ventral striatum, with repeated use, and as habit develops, there appears to be a shift from the drug to the conditioned stimulus. According to studies in laboratory animals, glutamatergic projections from prefrontal cortex and from amygdala into VTA/SN and NAc mediate these conditioned responses (Kalivas 2009). In this manner, the mere prediction of a reward may eventually become the reward that motivates the behaviour necessary for drug (or food) consumption.
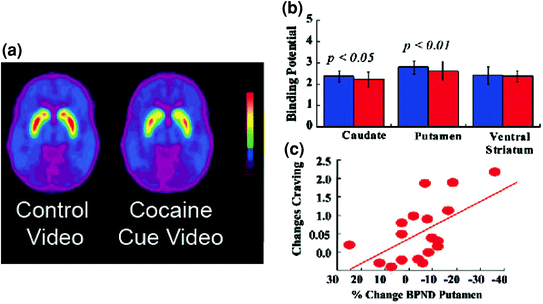
Fig. 5 DA changes induced by conditioned cues in active cocaine-addicted subjects. a Average nondisplaceable binding potential (BPND) images of [11C]raclopride in cocaine-addicted subjects (n = 17) tested while viewing a neutral video (nature scenes) and while viewing a cocaine-cues video (subjects administering cocaine). b D2R availability (BPND) in caudate, putamen, and ventral striatum for the neutral video (blue) and the cocaine-cues video (red). The cocaine cues decreased D2R in caudate and putamen. c Correlations between changes in D2R (reflecting DA increases) and self-reports of cocaine craving induced by the cocaine-cues video. Modified from ref. (Volkow et al. 2006b)
Interestingly, this type of functional “switch” has also been reported for natural reinforcers, which are likely to induce an equivalent and gradual shift in DA increases, from ventral to more dorsal regions of the striatum during the transition from a novel stimulus that is inherently rewarding to that of the associated cues that predict it. This transition is conveyed through DA signaling, which appears to code for a “reward prediction error” (Schultz 2010). The extensive glutamatergic afferents to DA neurons from regions involved in the processing of sensory (insula or primary gustatory cortex), homeostatic (hypothalamus), reward (NAc), emotional (amygdala and hippocampus), and multimodal (orbitofrontal cortex for salience attribution) information, modulate their activity in response to rewards and to conditioned cues (Geisler and Wise 2008). More specifically, projections from the amygdala and the orbitofrontal cortex (OFC) to DA neurons and to NAc are involved in conditioned responses to food (Petrovich 2010). Indeed, imaging studies showed that when non-obese male subjects were asked to inhibit their craving for food -while being exposed to food cues-, they exhibited decreased metabolic activity in amygdala and OFC (as well as in hippocampus), insula and striatum, and that the decreases in OFC were associated with reductions in food craving (Wang et al. 2009). A similar inhibition of the metabolic activity in the OFC (and also in NAc) has been observed in cocaine abusers when they were asked to inhibit their drug craving upon exposure to cocaine-cues (Volkow et al. 2009b).
Still, the emergence of such powerfully cue-conditioned cravings, which for food also occur in healthy individuals who do not overeat, would not be as devastating were they not coupled with growing deficits in the brain’s ability to inhibit maladaptive behaviours.
4 The Impact of Dysfunction in Inhibitory Control
The capacity to inhibit prepotent responses is bound to contribute to an individual’s ability to avoid engaging in inappropriate behaviours, such as taking drugs or eating past the point of satiety, and thus increasing his/her vulnerability to addiction (or obesity) (Volkow and Fowler 2000; Volkow et al. 2008a).
PET studies have uncovered significant reductions in D2R availability in the striatum of addicted subjects that persist for months after protracted detoxification [reviewed in (Volkow et al. 2009a)]. Similarly, preclinical studies in rodent and non-human primates have shown that repeated drug exposures are associated with reductions in striatal D2R levels (Nader et al. 2006; Thanos et al. 2007; Volkow et al. 2001). In the striatum, D2Rs mediate signaling in the striatal indirect pathway that modulates prefrontal regions; and its downregulation has been shown to enhance sensitization to the effects of drugs in animal models (Ferguson et al. 2011). In humans addicted to drugs, the reduction in striatal D2R is associated with decreased activity of prefrontal regions as evidenced by decreases in baseline glucose metabolism (a marker of brain function) in OFC, anterior cingulate gyrus (ACC), and dorsolateral prefrontal cortex (DLPFC) (Volkow et al. 2001, 1993, 2007) (Fig. 6). Inasmuch as OFC, ACC, and DLPFC are involved with salience attribution, inhibitory control/emotion regulation, and decision-making, respectively, it has been postulated that their improper regulation by D2R-mediated DA signaling in addicted subjects could underlie the enhanced motivational value of drugs in their behaviour and the loss of control over drug intake (Volkow and Fowler 2000). In addition, because impairments in OFC and ACC are associated with compulsive behaviours and impulsivity (Fineberg et al. 2009), DA’s impaired modulation of these regions is likely to contribute to the compulsive and impulsive drug intake seen in addiction (Goldstein and Volkow 2002). Indeed, in methamphetamine abusers, low striatal D2R was associated with impulsivity (Lee et al. 2009), and it also predicted compulsive cocaine administration in rodents (Everitt et al. 2008). A reverse scenario, in which an initial vulnerability for drug use preexists in prefrontal regions, and whereby repeated drug use triggers further decreases in striatal D2R, is also possible. Indeed, a study done in subjects who, despite having a high risk for alcoholism (positive family history of alcoholism) were not alcoholics, revealed a higher than normal striatal D2R availability that was associated with normal metabolism in OFC, ACC, and DLPFC (Volkow et al. 2006a). This suggests that, in these subjects at risk for alcoholism, the normal prefrontal function was linked to enhanced striatal D2R signaling, which in turn may have protected them from alcohol abuse.
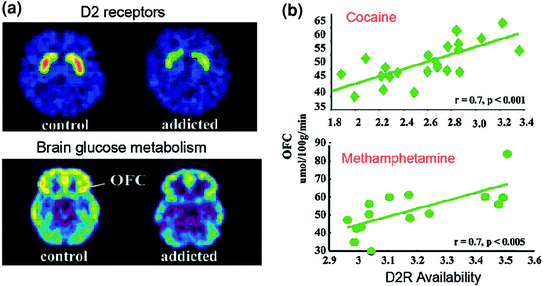
Fig. 6 Correlations between striatal D2R availability and metabolism in prefrontal brain regions. a Axial brain images for a control and for a cocaine-addicted subject for baseline images of D2R availability in striatum (obtained with [11C]raclopride) and of brain glucose metabolism in OFC (obtained with [18FDG). b Correlations between striatal D2R and metabolism in OFC in cocaine-addicted and methamphetamine-addicted subjects. Reprinted from Volkow et al. (2009a) Copyright (2009), with permission from Elsevier
Predictably, evidence of dysregulation in control circuits has also been found among obese individuals. Both preclinical and clinical studies have provided evidence of decreased striatal D2R signaling, which, as mentioned above, is linked with reward (NAc) but also with the establishment of habits and routines (dorsal striatum) in obesity (Geiger et al. 2009; Wang et al. 2001). Importantly, decreased striatal D2R availability has been linked to compulsive food intake in obese rodents (Johnson and Kenny 2010) and with decreased metabolic activity in OFC and ACC in obese humans (Volkow et al. 2008b) (Fig. 7a–c). Given that dysfunction in OFC and ACC results in compulsivity [see review (Fineberg et al. 2009)], this might be part of the mechanism by which low striatal D2R signaling facilitates hyperphagia (Davis et al. 2009). In addition, since decreased D2R-related signaling is also likely to reduce the sensitivity to other natural rewards, this deficit in obese individuals might also contribute to compensatory overeating (Geiger et al. 2008).
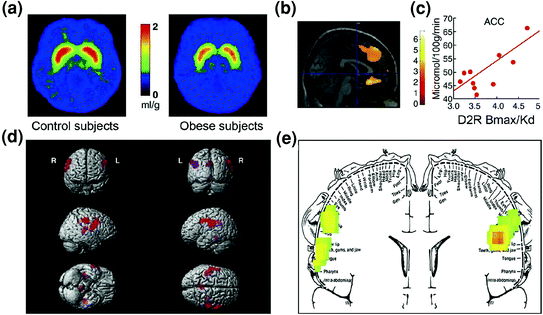
Fig. 7 Hyperphagia could result from a drive to compensate for a weakened reward circuit (processed through dopamine regulated corticostriatal circuits) combined with a heightened sensitivity to palatability (hedonic properties of food processed in part through the somatosensory cortex). a Averaged images for DA D2 receptor (D2R) availability in controls (n = 10) and in morbidly obese subjects (n = 10). b Results from (Statistical Parametric Mapping) SPM identifying the areas in the brain where D2R was associated with glucose metabolism, these included the medial OFC, ACC, and the dorsolateral PFC (region not shown). c Regression slope between striatal D2R and metabolic activity in ACC in obese subjects. d Three-dimensionally rendered SPM images showing the areas with higher metabolism in obese than in lean subjects (P < 0.003, uncorrected). e Colour coded SPM results displayed in a coronal plane with a superimposed diagram of the somatosensory homunculus. The results (z value) are presented using the rainbow scale where red > yellow > green. When compared with lean subjects, obese subjects had higher baseline metabolism in the somatosensory areas where the mouth, lips, and tongue are represented and which are involved with processing food palatability. Modified, with permission, from Volkow et al. (2008a) (a–c) and Wang et al. (2002) (d, e)
This hypothesis is consistent with preclinical evidence showing that decreased DA activity in VTA results in a dramatic increase in the consumption of high-fat foods (Stoeckel et al. 2008). Similarly, compared with normal-weight individuals, obese individuals who were presented with pictures of high-calorie food (stimuli to which they are conditioned) showed increased neural activation in regions that are part of reward and motivation circuits (NAc, dorsal striatum, OFC, ACC, amygdala, hippocampus, and insula) (Killgore and Yurgelun-Todd 2005). By contrast, in normal-weight controls, the activation of the ACC and OFC (regions involved in salience attribution that project into the NAc) during presentation of high-calorie food was found to be negatively correlated with their body mass index (BMI) (Stice et al. 2008b). This suggests a dynamic interaction between the amount of food eaten (reflected in part in the BMI) and the reactivity of reward regions to high-calorie food (reflected in the activation of OFC and ACC) in normal-weight individuals, which is lost in obesity.
Surprisingly, obese individuals exhibited less activation of reward circuits from actual food consumption (referred to as consummatory food reward) than lean individuals, whereas they showed greater activation of somatosensory cortical regions that process palatability when they anticipated consumption (Stice et al. 2008b). The latter observation corresponded to regions where a previous study had revealed enhanced activity in obese subject tested at baseline (non stimulation) (Wang et al. 2002) (Fig. 7d, e). An enhanced activity of regions that process palatability could make obese subjects favour food over other natural reinforcers, whereas decreased activation of dopaminergic targets by the actual food consumption might lead to overconsumption as a means to compensate for weak D2R-mediated signaling (Stice et al. 2008a). This reduced response of the reward circuitry to food consumption in obese subjects is reminiscent of the reduced DA increases triggered by drug consumption in addicted individuals when compared to non-addicted subjects.
The prefrontal cortex (PFC) plays a crucial role in executive function, including inhibitory control (Miller and Cohen 2001). These processes are modulated by D1R and D2R (presumably also D4R) and thus, the decreased activity in PFC, both in addiction and in obesity, is likely to contribute to poor control and high compulsivity. The lower-than-normal availability of D2R in the striatum of obese individuals, which has been associated with reduced activity in PFC and ACC (Volkow et al. 2008b) is therefore likely to contribute to their deficient control over food intake. Indeed, the negative correlation between BMI and striatal D2R reported in obese (Wang et al. 2001) and in overweight (Haltia et al. 2007a) individuals supports this. A better understanding of the mechanisms that lead to impaired PFC function in obesity (or addiction) could facilitate the development of strategies to ameliorate, or perhaps even reverse, specific impairments in crucial cognitive domains. For example, delay discounting, which is the tendency to devalue a reward as a function of the temporal delay of its delivery, is one of the most extensively investigated cognitive operations in relation to disorders associated with impulsivity and compulsivity. Delay discounting has been most exhaustively investigated in drug abusers who exhibit an exaggerated preference for small-but-immediate over large-but-delayed rewards (Bickel et al. 2007). However, the few studies performed with obese individuals have also uncovered evidence of a preference for high, immediate rewards, despite an increased chance of suffering higher future losses (Brogan et al. 2010; Weller et al. 2008). And more recently, another study found a positive correlation between BMI and hyperbolic discounting, whereby future negative payoffs are discounted less than future positive payoffs (Ikeda et al. 2010). Interestingly, delay discounting seems to depend on the function of ventral striatum (Gregorios-Pippas et al. 2009) and of the PFC, including lateral OFC (Bjork et al. 2009), and is sensitive to DA manipulations (Pine et al. 2010). Specifically, enhancing DA signaling (with L DOPA treatment) increased impulsivity and temporal discounting.
5 Involvement of Motivation Circuits
Dopaminergic signaling also modulates motivation. Behavioural traits such as vigor, persistence, and investing a continued effort towards achieving a goal, are all subject to modulation by DA acting through several target regions, including NAc, ACC, OFC, DLPFC, amygdala, dorsal striatum, and ventral pallidum (Salamone et al. 2007). Dysregulated DA signaling is associated with enhanced motivation to procure drugs, a hallmark of addiction, which is why drug-addicted individuals often engage in extreme behaviours to obtain drugs, even when they entail known severe and adverse consequences (Volkow and Li 2005). Because drug taking becomes the main motivational drive in drug addiction (Volkow et al. 2003), addicted subjects are aroused and motivated by the process of obtaining the drug but tend to become withdrawn and apathetic when exposed to non-drug-related activities. This shift has been studied by comparing the brain activation patterns occurring with exposure to conditioned cues with those occurring in the absence of such cues. In contrast to the decreases in prefrontal activity reported in detoxified cocaine abusers when not stimulated with drug or drug cues [see review (Volkow et al. 2009a)], these prefrontal regions become activated when cocaine abusers are exposed to craving-inducing stimuli (either drugs or cues) (Grant et al. 1996; Volkow et al. 1999a; Wang et al. 1999). This result is reminiscent of the observation that cocaine abusers, studied shortly after an episode of cocaine binging, showed an increase in metabolic activity in OFC and ACC (also dorsal striatum) that was associated with craving (Volkow et al. 1991).
Moreover, when the responses to i.v. MP are compared between cocaine-addicted and non-addicted individuals, the former responded with increased metabolism in ventral ACC and medial OFC (an effect associated with craving), while the latter showed the opposite response, namely decreased metabolism in these regions (Volkow et al. 2005). This suggests that the activation of these prefrontal regions with drug exposure may be specific to addiction and associated with the enhanced desire for the drug. In addition, a study that prompted cocaine-addicted subjects to purposefully inhibit craving when exposed to drug cues showed that those subjects who were successful at inhibiting craving displayed decreased metabolism in medial OFC (which processes motivational value of a reinforcer) and NAc (which predicts reward) (Volkow et al. 2009b). These findings further corroborate the involvement of OFC, ACC, and striatum in the enhanced motivation to procure the drug seen in addiction.
Predictably, the OFC has also been implicated in attributing salience value to food (Grabenhorst et al. 2008; Rolls and McCabe 2007), helping to assess its expected pleasantness and palatability as a function of its context. PET studies with FDG to measure brain glucose metabolism in normal weight individuals reported that exposure to food-cues increased metabolic activity in OFC, which was an effect associated with the perception of hunger and the desire for food (Wang et al. 2004). The enhanced OFC activation by the food stimulation is likely to reflect downstream dopaminergic effects and participate in DA’s involvement in the drive for food consumption. The OFC plays a role in learning stimulus-reinforcement associations and conditioning (Cox et al. 2005; Gallagher et al. 1999), supports conditioned-cue elicited feeding (Weingarten 1983), and probably contributes to overeating irrespective of hunger signals (Ogden and Wardle 1990). Indeed, dysfunction of the OFC has been linked to overeating (Machado and Bachevalier 2007).
In spite of some inconsistencies among studies, brain imaging data also support the notion that structural and functional changes in brain regions implicated in executive function (including inhibitory control) may be associated with high BMI in otherwise healthy individuals. For example, an MRI study done in elderly women, using voxel-based morphometry, found a negative correlation between BMI and gray matter volumes (including frontal regions), which, in the OFC, was associated with impaired executive function (Walther et al. 2010). Using PET to measure brain glucose metabolism in healthy controls, we reported a negative correlation between BMI and metabolic activity in DLPFC, OFC, and ACC. In this study, the metabolic activity in prefrontal regions predicted the subjects’ performance in tests of executive function (Volkow et al. 2009c). Similarly, a nuclear magnetic resonance (NMR) spectroscopic study in healthy middle age and elderly controls showed that BMI was negatively associated with the levels of N-acetyl-aspartate (a marker of neuronal integrity) in frontal cortex and ACC (Gazdzinski et al. 2008; Volkow et al. 2009c).
Brain imaging studies comparing obese and lean individuals have also reported lower gray matter density in frontal regions (frontal operculum and middle frontal gyrus) and in post-central gyrus and putamen (Pannacciulli et al. 2006). Another study, found no differences in gray matter volumes between obese and lean subjects, however, it did record a positive correlation between white matter volume in basal brain structures and waist to hip ratios, a trend that was partially reversed by dieting (Haltia et al. 2007b). Interestingly, cortical areas, like the DPFC and OFC that are involved in inhibitory control, have also been found to become activated in successful dieters in response to meal consumption (DelParigi et al. 2007), suggesting a potential target for behavioural retraining in the treatment of obesity (and also in addiction).
6 Involvement of Interoceptive Circuitry
Neuroimaging studies have revealed that the middle insula plays a critical role in cravings for food, cocaine, and cigarettes (Bonson et al. 2002; Pelchat et al. 2004; Wang et al. 2007). The importance of the insula has been highlighted by a study that reported that smokers with damage to this region (but not control smokers who had suffered extra-insular lesions) were able to stop smoking easily and without experiencing either cravings or relapse (Naqvi et al. 2007). The insula, particularly its more anterior regions, is reciprocally connected to several limbic regions (e.g., ventromedial prefrontal cortex, amygdala, and ventral striatum) and appears to have an interoceptive function, integrating the autonomic and visceral information with emotion and motivation, thus providing conscious awareness of these urges (Naqvi and Bechara 2009). Indeed, brain lesion studies suggest that the ventromedial PFC and insula are necessary components of the distributed circuits that support emotional decision-making (Clark et al. 2008). Consistent with this hypothesis, imaging studies consistently show differential activation of the insula during craving (Brody et al. 2009; Goudriaan et al. 2010; Naqvi and Bechara 2009; Wang et al. 1999). Accordingly, the reactivity of this brain region has been suggested to serve as a biomarker to help predict relapse (Janes et al. 2010).
The insula is also a primary gustatory area, which participates in many aspects of eating behaviours, such as taste. In addition, the rostral insula (connected to primary taste cortex) provides information to the OFC that influences its multimodal representation of the pleasantness or reward value of incoming food (Rolls 2008). Because of the involvement of the insula in the interoceptive sense of the body, in emotional awareness (Craig 2003) and in motivation and emotion (Rolls 2008), a contribution of insular impairment in obesity could be expected. Indeed, gastric distention results in activation of the posterior insula, which is likely to reflect its role in the awareness of body states (in this case of fullness) (Wang et al. 2008). Moreover, in lean, but not in obese subjects, gastric distention resulted in activation of the amygdala and deactivation of the anterior insula (Tomasi et al. 2009). The lack of amygdala response in obese subjects could reflect a blunted interoceptive awareness of bodily states linked with satiety (full stomach). Even though the modulation of insular activity by DA has been poorly investigated, it is recognised that DA is involved in the responses to tasting of palatable foods that are mediated through the insula (Hajnal and Norgren 2005). Human imaging studies have shown that tasting palatable foods activated the insula and midbrain areas (DelParigi et al. 2005; Frank et al. 2008). However, the DA signaling may also be necessary for sensing the calorie content of food. For example, when normal weight women tasted a sweetener with calories (sucrose), both the insula and dopaminergic midbrain areas became activated, whereas tasting a calorie-free sweetener (sucralose) only activated the insula (Frank et al. 2008). Obese subjects exhibit greater insular activation than normal controls when tasting a liquid meal that consists of sugar and fat (DelParigi et al. 2005). In contrast, subjects who have recovered from anorexia nervosa show less activation in the insula when tasting sucrose and no association of feelings of pleasantness with insular activation as observed in the normal controls (Wagner et al. 2008). When combined, these results make it likely that dysregulation of the insula in response to taste stimuli might be involved in the impaired control of various appetitive behaviours.
7 The Circuitry of Aversion
As mentioned before, training (conditioning) on a cue that predicts reward leads to dopaminergic cells firing in response to the prediction of reward, and not to the reward itself. On the other hand, and consistent with this logic, it has been observed that dopaminergic cells will fire less than normal if the expected reward fails to materialise (Schultz et al. 1997). Cumulative evidence (Christoph et al. 1986; Lisoprawski et al. 1980; Matsumoto and Hikosaka 2007; Nishikawa et al. 1986) points to the habenula as one of the regions that controls the decreases in firing of dopaminergic cells in VTA that may follow the failure to receive an expected reward (Kimura et al. 2007). Thus, an enhanced sensitivity of the habenula, as a result of chronic drug exposures, could underlie a greater reactivity to drug cues. Indeed, activation of the habenula, in cocaine-addicted subjects, has been associated with behavioural relapse to drug taking upon cue exposure (Brown et al. 2011; Zhang et al. 2005). In the case of nicotine, the α5 nicotinic receptors in the habenula appear to modulate the aversive responses to large doses of nicotine (Fowler et al. 2011); and the α5 and α2 receptors in the habenula are implicated in nicotine withdrawal (Salas et al. 2009). Because of the habenula’s opposite response to that of DA neurons to reward (deactivation) and its activation upon exposure to aversive stimuli, we refer here to the habenula signaling as one conveying an “antireward” input.
The habenula appears to play a similar role with regards to food reward. A highly palatable food diet can induce obesity in rats, with the weight increases correlating with increases in μ-opioid peptide binding in the basolateral and basomedial amygdala. Interestingly, the medial habenula showed significantly higher μ-opioid peptide binding (by approximately 40%) after exposure to the palatable food in the rats that gained weight (those that consumed more food) but not in those that did not (Smith et al. 2002). This suggests that the habenula may be involved in overeating under conditions of availability of palatable food. Moreover, neurons in the rostromedial tegmental nucleus, which receive a major input from the lateral habenula, project to VTA DA neurons and are activated after food deprivation (Jhou et al. 2009). These findings are consistent with a role for the habenula in mediating responses to aversive stimuli or states such as those that occur during dieting or drug withdrawal.
The involvement of the habenula as an antireward hub within emotional networks is consistent with prior theoretical models of addiction that postulated sensitized anti-reward responses (mediated through enhanced sensitivity of the amygdala and increased signaling though the corticotropin releasing factor) as driving drug intake in addiction (Koob and Le Moal 2008). Similar antireward responses may also contribute to excessive food consumption in obesity.
8 Pathological Drug and Food Reward: An Updated Working Model
The ability to resist the urge to use a drug or eat past the point of satiety requires the proper functioning of neuronal circuits involved in top-down control to oppose the conditioned responses that predict reward from ingesting the food/drug and the desire to ingest the food/drug. Here, we highlighted six of these circuits: reward/saliency, conditioning/habits, inhibitory control/executive function, motivation/drive, interoception, and aversion avoidance/stress reactivity (Fig. 8). Based on the imaging data presented here, we postulate that it is the discrepancy between the expectation for the drug/food effects (conditioned responses) and the blunted neurophysiological effects that maintains the taking of drugs or the overconsumption of foods in an attempt to attain the expected reward. Also, whether tested during early or protracted periods of abstinence/dieting, addicted/obese subjects show lower D2R in striatum (including NAc), which are associated with decreases in baseline activity in frontal brain regions implicated in salience attribution (orbitofrontal cortex) and inhibitory control (ACC and DLPFC), whose disruption results in compulsivity and impulsivity. Finally, evidence has also been emerging on the role of interoceptive and aversive circuitry in the systemic imbalances that result in the compulsive consumption of either drugs or food.
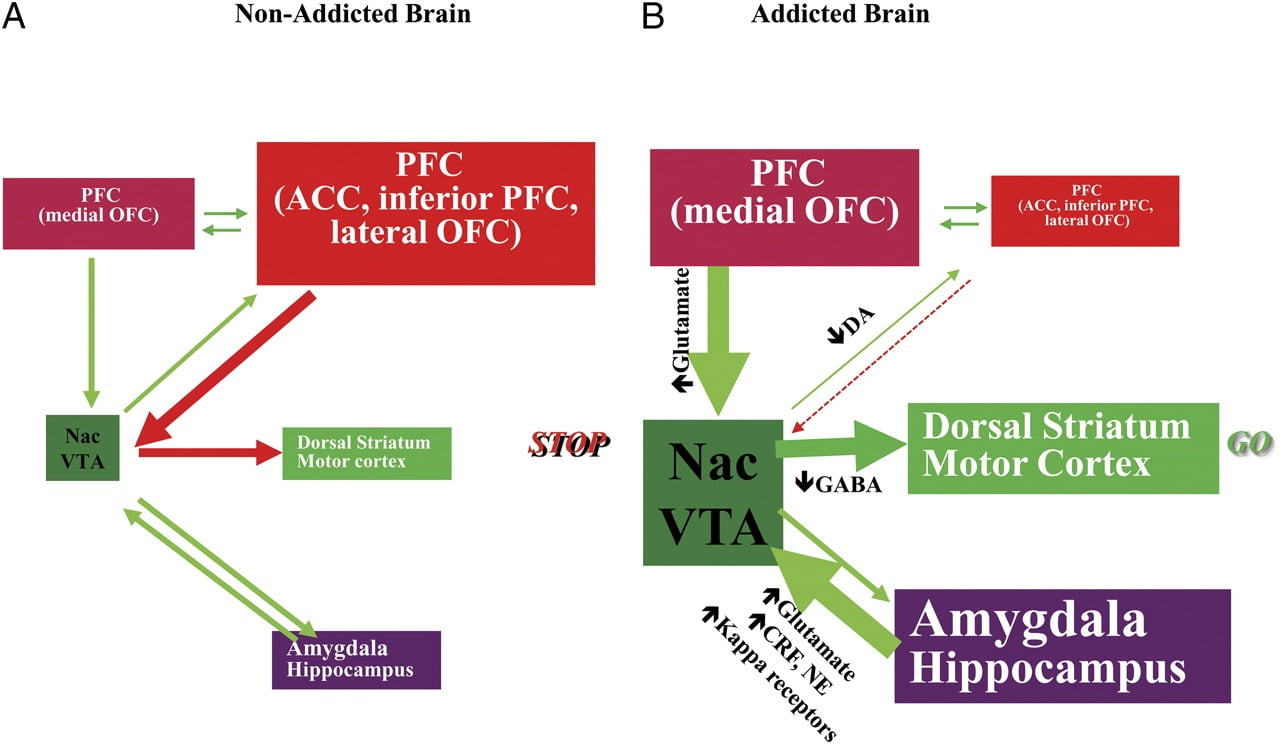
Fig. 8 Model proposing a network of interacting circuits, disruptions which contribute to the complex set of stereotypic behaviours underlying drug addiction and chronic overeating: reward (nucleus accumbens, VTA, and ventral pallidum), conditioning/memory (amygdala, medial OFC for attribution of saliency, hippocampus, and dorsal striatum for habits), executive control (DLPFC, ACC, inferior frontal cortex, and lateral OFC), motivation/drive (medial OFC for attribution of saliency, ventral ACC, VTA, SN, dorsal striatum, and motor cortex). Nac, nucleus accumbens, interoception (Insula and ACC), and aversion/avoidance (Habenula). a When these circuits are balanced, this results in proper inhibitory control and decision making. b During addiction, when the enhanced expectation value of the drug in the reward, motivation, and memory circuits overcomes the control circuit, favouring a positive-feedback loop initiated by the consumption of the drug and perpetuated by the enhanced activation of the motivation/drive and memory circuits. These circuits also interact with circuits involved in mood regulation, including stress reactivity (which involves the amygdala, hypothalamus, habenula) and interoception (which involves the insula and ACC and contributes to awareness of craving). Several neurotransmitters are implicated in these neuroadaptations, including glutamate, GABA, norepinephrine, corticotropin-releasing factor, and opioid receptors. CRF, corticotropin-releasing factor; NE, norepinephrine. Modified with permission from Volkow et al. (2011b)
As a consequence of the sequential disruption in these circuits, individuals may experience 1) an enhanced motivational value of the drug/food (secondary to learned associations through conditioning and habits) at the expense of other reinforcers (secondary to decreased sensitivity of the reward circuit), 2) an impaired ability to inhibit the intentional (goal-directed) actions triggered by the strong desire to take the drug/food (secondary to impaired executive function) that result in compulsive drug/food taking, and 3) enhanced stress reactivity and aversive avoidance that results in impulsive drug taking to escape the aversive state.
This model suggests a multipronged therapeutic approach to addiction designed to decrease the reinforcing properties of drug/food, reestablish/enhance the rewarding properties of natural reinforcers, inhibit conditioned learned associations, enhance motivation for non-drug/food-related activities, decrease stress reactivity, improve mood, and strengthen general-purpose inhibitory control.
Acknowledgments
The authors would like to thank the support of the NIAAA intramural program of the National Institutes of Health.
References