COMMENTS: a review by the top researcher on obesity and food addiction.
Volume 69, Issue 4, 24 February 2011, Pages 664–679
http://dx.doi.org/10.1016/j.neuron.2011.02.016,
Review
Paul J. Kenny1, ,
1 Laboratory of Behavioral and Molecular Neuroscience, Department of Molecular Therapeutics, The Scripps Research Institute, Jupiter, FL 33458, USA
________________________________________
Food is consumed in order to maintain energy balance at homeostatic levels. In addition, palatable food is also consumed for its hedonic properties independent of energy status. Such reward-related consumption can result in caloric intake exceeding requirements and is considered a major culprit in the rapidly increasing rates of obesity in developed countries. Compared with homeostatic mechanisms of feeding, much less is known about how hedonic systems in brain influence food intake. Intriguingly, excessive consumption of palatable food can trigger neuroadaptive responses in brain reward circuitries similar to drugs of abuse. Furthermore, similar genetic vulnerabilities in brain reward systems can increase predisposition to drug addiction and obesity. Here, recent advances in our understanding of the brain circuitries that regulate hedonic aspects of feeding behavior will be reviewed. Also, emerging evidence suggesting that obesity and drug addiction may share common hedonic mechanisms will also be considered.
________________________________________
Main Text
“There is no sincerer love than the love of food.”
—George Bernard Shaw
Introduction
Obesity, defined as a body mass index (BMIs) of >30, is a condition in which adiposity is abnormally high and can result from hyperphagia or decreased metabolic rate (O’Rahilly, 2009). Excessive adiposity is a major risk factor for cardiovascular disease, cancer, type 2 diabetes, and mood-related disorders, with obese individuals often suffering social stigmatization ( [Bean et al., 2008], [Centers for Disease Control and Prevention, 2009] and [Luppino et al., 2010]). According to the Center for Disease Control (CDC), obesity-related health care expenses in the United States between 1998 and 2000 were approximately $213 billion. Further, 300,000 deaths in the United States each year can be attributed to overweight- and obesity-related diseases (Allison et al., 1999), with obesity the second leading cause of preventable death behind tobacco use. Nevertheless, the prevalence of obesity in Western societies continues to increase dramatically, with current estimates suggesting that greater than 30% of adults in the United States are obese (Flegal et al., 2010).
Most conceptualizations of feeding regulation propose that two parallel systems interact to influence food intake ( [Hommel et al., 2006], [Lutter and Nestler, 2009] and [Morton et al., 2006]). The homeostatic system comprises hormonal regulators of hunger, satiety, and adiposity levels, such as leptin, ghrelin, and insulin, which act on hypothalamic and brainstem circuits to stimulate or inhibit feeding in order to maintain appropriate levels of energy balance. Dysfunction in components of the homeostatic system, such as congenital leptin deficiency, can result in a persistent state of positive energy balance and the development of obesity ( [Campfield et al., 1995], [Halaas et al., 1995] and [Pelleymounter et al., 1995]). The mechanisms through which hormonal regulators of hunger and satiety act on hypothalamic and brain stem circuitries to maintain energy homeostasis have been described in detail elsewhere, and readers interested in this topic are referred to the many excellent reviews on this subject (for example, [Abizaid et al., 2006a] and [Gao and Horvath, 2007]).
In addition to metabolic systems, brain reward systems also play an important role in feeding behavior ( [Lutter and Nestler, 2009] and [Saper et al., 2002]). In general, bland tasting foods are not eaten to excess, whereas palatable foods are often consumed even after energy requirements have been met. Ease of access to palatable energy-dense food is considered a major environmental risk factor for obesity (Volkow and Wise, 2005), and overconsumption of palatable food is considered a major factor contributing to the recent surge in obesity ( [Finkelstein et al., 2005], [Hill et al., 2003] and [Swinburn et al., 2009]). Indeed, obtaining the pleasurable effects of palatable food is a powerful motivating force that in certain individuals can override homeostatic signals ( [Shomaker et al., 2010], [Sunday et al., 1983] and [Zheng et al., 2009]). When presented with a choice, rats overwhelmingly prefer to consume a calorie-free saccharin solution rather than self-administer intravenous cocaine infusions (Lenoir et al., 2007). Moreover, well-fed rats will voluntarily expose themselves to extreme cold (−15°C), noxious heat pain or aversive footshock to obtain palatable food items, such as shortcake, meat pâté, peanut butter, Coca-Cola, M&M candies, chocolate chips, or yogurt drops, even when less palatable standard chow is freely available ( [Cabanac and Johnson, 1983], [Foo and Mason, 2005] and [Oswald et al., 2010]). These findings highlight just how intensely macronutrients in palatable food can stimulate brain reward systems independent of their caloric value ( [Wang et al., 2004a] and [Wang et al., 2004b]) and how high the motivation to consume palatable food can be even in the absence of homeostatic energy requirements. Drugs of abuse such as cocaine or nicotine can similarly induce high levels of consummatory behavior even though they are devoid of caloric or nutrient value. In fact, because of the many similarities between overeating in obesity and excessive drug use in addiction (Volkow and Wise, 2005), it has been argued that obesity should be considered as a brain disorder and included as a diagnostic category in the forthcoming fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) ( [Devlin, 2007] and [Volkow and O’Brien, 2007]). Compared with homeostatic mechanisms of feeding behavior much less is known about precisely how hedonic systems influence food intake. Similarly, the influence of intrinsic or diet-induced alterations on the responsiveness of brain reward systems, and how these effects contribute to overeating and obesity, remains unclear. Summarized below are recent data highlighting advances in our understanding of hedonic mechanisms of eating and diet-induced alterations in brain reward activity that may contribute to the development of obesity
Activation of Brain Reward Systems in Response to Palatable Food: Interactions with Hormonal Regulators of Energy Balance
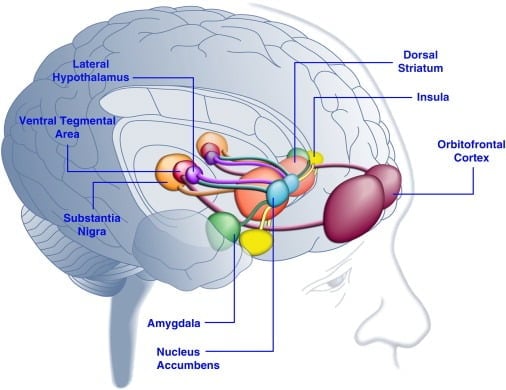
Consumption of palatable food can enhance mood in humans ( [Dallman et al., 2003] and [Macht and Mueller, 2007]) and support the establishment of a conditioned place preference in laboratory animals ( [Imaizumi et al., 2001] and [Sclafani et al., 1998]). These effects are likely related to the stimulation of brain reward systems by palatable food (Figure 1). Indeed, human brain imaging studies have shown that food and food–related visual or olfactory cues can activate corticolimbic and meso accumbens brain circuits implicated in reward, most notably the orbitofrontal cortex (OFC), insula, amygdala, hypothalamus, striatum, and midbrain regions including the ventral tegmental area (VTA) and substantia nigra (SN) ( [Bragulat et al., 2010], [Pelchat et al., 2004], [Schur et al., 2009] and [Simmons et al., 2005]). The striatum, insula, anterior cingulate cortex, and midbrain structures encode the subjective value of rewards regardless of their type (e.g., food, sex, monetary rewards), consistent with a role for this neuronal network in general hedonic representation (Sescousse et al., 2010). In contrast, the OFC appears to play a particularly prominent role in representations related to the value of specific types of rewards including palatable food ( [Man et al., 2009], [Rolls, 2008] and [Sescousse et al., 2010]). Hunger can enhance palatable food-induced activation of corticolimbic and midbrain regions in humans (LaBar et al., 2001). For example, the intensity of activation of the ventral striatum, amygdala, insula, and OFC in response to high-calorie palatable food was far greater when human subjects were hungry rather than well fed (Goldstone et al., 2009). This is consistent with the fact that periods of hunger and dieting increase self-reported ratings of the “power” of palatable food and craving for “tempting” food ( [Hofmann et al., 2010] and [Rolls et al., 1983]). Conversely, overfeeding can reduce neuronal responses to palatable food, particularly in the insular cortex and hypothalamus (Cornier et al., 2009). Hence, the hedonic value of food is influenced by metabolic state, suggesting that regulators of metabolism such as leptin and ghrelin may influence the activity of hedonic systems in the brain. Consistent with this view, human subjects treated with leptin or the gut-derived postprandial factor peptide YY3-36 (PYY) ( [Batterham et al., 2007] and [Farooqi et al., 2007]), or those that underwent gastric distention mimicking meal ingestion (Wang et al., 2008), had reduced activity in reward-related brain regions. Conversely, hyperphagic human patients with congenital leptin deficiency demonstrate increased activity in the insular cortex and striatum in response to images of food ( [Baicy et al., 2007] and [Farooqi et al., 2007]). In these individuals, leptin replacement therapy attenuated the enhanced insular and striatal activity and decreased self-reported liking of food ( [Baicy et al., 2007] and [Farooqi et al., 2007]). Leptin treatment also blocks the rewarding properties of sucrose in food-restricted rats similar to the dopamine receptor antagonist α-flupenthixol (Figlewicz et al., 2001). Moreover, leptin receptors are expressed on midbrain dopamine neurons in the VTA and SN (Figlewicz et al., 2003), suggesting that leptin may influence hedonic aspects of feeding behavior through modulation of mesostriatal dopamine transmission. Confirming this possibility, leptin infusions into the VTA inhibited the activity of dopamine neurons and decreased food intake in rats (Hommel et al., 2006; see also Krügel et al., 2003). Conversely, knockdown of leptin receptors in the VTA increased food intake, enhanced locomotor activity, and increased preference for palatable food in rats (Hommel et al., 2006). Leptin therefore exerts an inhibitory influence on mesoaccumbens dopamine transmission, a neurotransmitter system that has been heavily implicated in reward and motivation but less so in energy homeostasis ( [de Araujo et al., 2010] and [Vucetic and Reyes, 2010]). More recently, the hunger-related hormone ghrelin ( [Kojima et al., 1999] and [Nakazato et al., 2001]) was shown to potentiate the activation of hedonic systems in the brain in response to food cues (Malik et al., 2008). Specifically, ghrelin enhanced the activation of OFC, amygdala, insula, striatum, VTA, and SN in response to pictures of highly palatable food in obese individuals (Malik et al., 2008). In rats, ghrelin exerts a stimulatory effect on midbrain dopamine systems ( [Abizaid et al., 2006b], [Jerlhag et al., 2006] and [Jerlhag et al., 2007]) and increases the rewarding value of palatable food (Perello et al., 2010). Taken together, these findings demonstrate that palatable food activates brain reward systems and that hormonal regulators of appetite can influence food intake in part by modulating hedonic responses to food.
Figure 1. Areas of the Human Brain Activated in Response to Palatable Food or Food-Associated Cues. The orbitofrontal cortex and amygdala are thought to encode information related to the reward value of food ( [Baxter and Murray, 2002], [Holland and Gallagher, 2004], [Kringelbach et al., 2003], [O’Doherty et al., 2002] and [Rolls, 2010]). The insula processes information related to the taste of food and its hedonic valuation ( [Balleine and Dickinson, 2000] and [Small, 2010]). The nucleus accumbens and dorsal striatum, which receive dopaminergic input from the ventral tegmental area and substantia nigra, regulate the motivational and incentive properties of food ( [Baicy et al., 2007], [Berridge, 1996], [Berridge, 2009], [Farooqi et al., 2007], [Malik et al., 2008] and [Söderpalm and Berridge, 2000]). The lateral hypothalamus may regulate rewarding responses to palatable food and drive food-seeking behaviors (Kelley et al., 1996). These brain structures act in a concerted manner to regulate learning about the hedonic properties of food, shifting attention and effort toward obtaining food rewards and regulating the incentive value of environmental stimuli that predict availability of food rewards (Dagher, 2009). For the sake of clarity, not all interconnections between these structures are shown.
Similar brain regions are activated by palatable food in the rat brain as those activated in humans, as measured by expression of immediate early genes (IEG) such as c-fos, arc, or zif268. Indeed, palatable food activates the dorsal and ventral striatum, VTA, lateral hypothalamus (LH), and central and basolateral nuclei of the amygdala and reward-related cortical structures in rats ( [Angeles-Castellanos et al., 2007], [Park and Carr, 1998] and [Schiltz et al., 2007]). Interestingly, Fos immunoreactivity was actually decreased in the lateral and medial habenula in rats after palatable food consumption (LHb) (Park and Carr, 1998). In nonhuman primates, the LHb is activated by aversive stimuli or omission of expected rewards and inhibited by the delivery of a palatable juice reward (Matsumoto and Hikosaka, 2007). In addition, LHb activity inhibits reward-related mesoaccumbens dopamine-containing neurons through an indirect pathway involving the rostromedial tegmental nucleus (RMTg) (Jhou et al., 2009). Habenular activity is therefore inversely related to food hedonics, suggesting that the habenular complex may influence nonhomeostatic eating. Indeed, activation of the LHb was recently shown to decrease sucrose consumption in rats, whereas lesions of LHb increase sucrose seeking behavior (Friedman et al., 2011). Considering that the habenular complex is small and challenging to identify and functionally image in humans (Salas et al., 2010), this may explain why alterations in habenular activity have not been reported in human imaging studies in response to palatable food.
Brain Circuitries that Regulate Hedonic Eating: Midbrain Dopamine Systems
The mesoaccumbens dopamine pathway is activated in humans and laboratory animals in response to palatable food or appetitive food-related cues and leptin, ghrelin, and other regulators of appetite influence activity in this system. This suggests that midbrain dopamine systems play an important role in palatable food consumption. Perhaps the clearest indication that midbrain dopamine transmission influences palatable food intake in humans is the fact that Parkinson’s disease (PD) patients, in which there is degeneration of dopamine-containing neurons in the midbrain, tend to consume less food than nonaffected individuals (Nirenberg and Waters, 2006). Moreover, treatment of PD patients with dopamine receptor agonists can trigger compulsive-like consumption of palatable food ( [Dagher and Robbins, 2009] and [Nirenberg and Waters, 2006]). In fact, dopamine receptor agonists can induce hedonic overeating even in non-PD individuals (Cornelius et al., 2010). In animals, palatable sucrose solutions stimulate dopamine transmission in the NAc (Hernandez and Hoebel, 1988), an effect consistent with human brain imaging studies (Small et al., 2003). Using fast-scan cyclic voltammetry, it was shown that cues predicting delivery of a sucrose reward or the unexpected delivery of sucrose evoked dopamine transmission in NAc ( [Roitman et al., 2004] and [Roitman et al., 2008]). Further, the unexpected delivery of noxious quinine solutions had the opposite effect, decreasing accumbal dopamine transmission (Roitman et al., 2008). Finally, mice in which the enzyme tyrosine hydroxylase (TH) has been inactivated, causing them to be dopamine deficient, still demonstrate a marked preference for sucrose (or saccharin) solutions compared with water but consume less total amounts of the sucrose than control mice (Cannon and Palmiter, 2003). This suggests that dopamine-deficient mice can still detect sucrose palatability and prefer these solutions to water but are not able to sustain consumption of palatable solutions. It has therefore been proposed that mesoaccumbens dopamine transmission regulates motivational aspects of feeding behavior that are involved in food procurement and that other neurotransmitter systems likely regulate hedonic aspects of palatable food consumption.
Brain Circuitries that Regulate Hedonic Eating: Striatohypothalamic Systems
Infusion of μ-opioid receptor agonists such as [D-Ala2-N-Me-Phe4-gly-ol5]-enkephalin (DAMGO) into the NAc stimulates feeding behavior in rats with ad libitum access to food (i.e., nonhomeostatic feeding) ( [Peciña and Berridge, 2005] and [Zhang et al., 1998]). Conversely, opioid receptor antagonists infused into the NAc decrease consumption of preferred food without affecting intake of less palatable alternatives (Kelley et al., 1996). These data are consistent with the view that striatal opioid systems regulate the hedonic properties of palatable food. The shell region of the NAc and in particular hedonic “hot spots” in the rostrodorsal region of the medial shell ( [Peciña and Berridge, 2005] and [Peciña et al., 2006b]) plays a particularly important role in nonhomeostatic feeding. Because μ-opioid receptor activation results in the inhibition of medium spiny neuron activity in the NAc, it has been proposed that the NAc shell exerts a tonic inhibitory influence on palatable food consumption (Kelley et al., 2005). Consistent with this view, stimulation of inhibitory GABAA or GABAB receptors ( [Basso and Kelley, 1999] and [Stratford and Kelley, 1997]) or blockade of excitatory ionotropic glutamate receptors (Maldonado-Irizarry et al., 1995) in the NAc shell increases food consumption. Similarly, excitotoxic lesion of the NAc shell also increases food consumption and enhances sensitivity to food reward ( [Johnson et al., 1996] and [Maldonado-Irizarry and Kelley, 1995]). In particular, consumption of energy-dense palatable food is preferentially triggered by these manipulations ( [Basso and Kelley, 1999], [Kelley et al., 2005] and [Zhang et al., 1998]).
Considering the major influence of accumbal signaling on hedonic feeding, Thompson and Swanson (2010) used a circuit tracing procedure to accurately identify the precise anatomical networks through which the NAc may influence palatable food consumption. In these elegant studies, rats received two nonoverlapping injections of anterograde/retrograde tracers (termed COINs) into sites of the NAc shell that powerfully influence palatable food consumption, and afferent/efferent connections were identified. It was shown that feeding-related sites in the NAc extend inhibitory projections predominately to the anterior LH and ventral pallidum (VP) (Thompson and Swanson, 2010). Unlike the rest of the NAc, which projects densely to the VTA, food-related hedonic hot spots in NAc shell project to the interfascicular nucleus (IFN), a structure located adjacent to the VTA that extends dopaminergic projections in a reciprocal manner back to the NAc shell (Thompson and Swanson, 2010). Further, the anterior LH projects to the LHb (Thompson and Swanson, 2010), again suggesting that the habenular complex may play a role in food hedonics (Friedman et al., 2011).
The above data show that the LH receives prominent inhibitory input from sites in NAc that exert a tonic inhibitory influence on palatable food consumption. The LH also has functional connectivity with other cortical and limbic brain sites implicated in organizing and directing behavior toward obtaining palatable food (Figure 1), such as the OFC, insula, and amygdala. Importantly, inactivation of the LH abolishes the stimulatory effects of NAc manipulations on food intake ( [Maldonado-Irizarry et al., 1995] and [Will et al., 2003]). Furthermore, inactivation of the NAc shell enhances the activity of the LH, particularly LH neurons that synthesize the neuropeptide hypocretin (also known as orexin), as measured by Fos immunoreactivity ( [Baldo et al., 2004] and [Stratford and Kelley, 1999]). Indeed, infusion of the μ-opioid receptor agonist DAMGO into the NAc shell activates hypocretin-containing neurons in the hypothalamus (Zheng et al., 2007), and disruption of hypocretin transmission in VTA abolishes palatable food intake triggered by intra-NAc DAMGO infusions (Zheng et al., 2007). Thus, hedonic hot spots in the NAc shell exert a tonic inhibitory influence on LH neurons, and in particular hypocretin-containing neurons (Louis et al., 2010), thereby limiting consumption of palatable food. Disruption of this accumbal “stop signal,” through enhanced opioid receptor signaling for example, results in enhanced LH activity that drives nonhomeostatic consumption of palatable food (Figure 2).
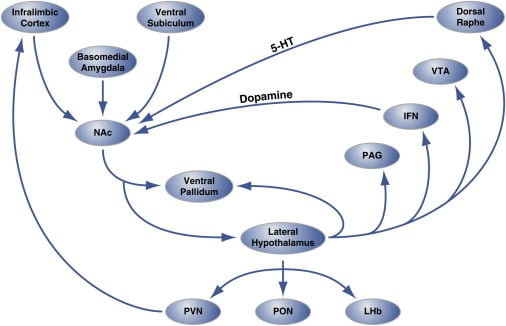
Figure 2. Circuit-Level Organization of Hedonic “Hot Spots” in Nucleus Accumbens Shell that Regulate Hedonic Eating
The shell region of the nucleus accumbens (NAc) receives innervation from cortical and limbic brain sites and projects to the lateral hypothalamus and ventral pallidum. In turn, the lateral hypothalamus also projects to the ventral pallidum and also the PAG, IFN, VTA, and dorsal raphe nucleus. The IFN and dorsal raphe extend dopaminergic and serotonergic projections, respectively, back to the NAc. The lateral hypothalamus also innervates thalamic (PVN and PON) and epithalamic (LHb) structures. Not shown are the minor projections from the lateral hypothalamus to septal brain areas. 5-HT, serotonin; IFN, interfascicular nucleus; LHb, lateral habenula; PON, preoptic nucleus; PVN, paraventricular nucleus of the thalamus; VTA, ventral tegmental area. Figure is adapted with permission from Thompson and Swanson (2010).
Brain Circuitries that Regulate Hedonic Eating: Striatopallidal Systems
In addition to the LH, NAc shell neurons also project to the VP (Figure 2). In an interesting series of experiments, it was shown that accumbal projections to the VP and LH may regulate dissociable aspects of nonhomeostatic eating (Smith and Berridge, 2007). DAMGO infusions into either the NAc shell or VP increased orofacial reactions to sucrose solutions hypothesized to reflect “liking” reactions in rats (i.e., palatability response) and also increased food consumption (Smith and Berridge, 2007). Infusions of naloxone into either NAc or VP decreased facial liking reactions to sucrose, suggesting that synchronized opioid transmission in NAc and VP is necessary to process information related food palatability. However, naloxone infused into the NAc, but not the VP, decreased nonhomeostatic eating (Smith and Berridge, 2007), suggesting that nonhomeostatic eating occurs independent of this NAc→VP connectivity and instead likely relies on the NAc→LH pathway ( [Smith and Berridge, 2007] and [Taha et al., 2009]). Consistent with the notion that aspects of nonhomeostatic eating can be dissociated, single-unit recordings have shown that a population of NAc neurons appears to selectively encode information related to the relative reinforcing properties of food (i.e., palatability) (Taha and Fields, 2005). In the same rats, changes in the activity a second population of NAc neurons appeared to coincide with the initiation of feeding behavior (Taha and Fields, 2005).
Brain Circuitries that Regulate Hedonic Eating: Amygdalar Systems
Further supporting the notion that aspects of nonhomeostatic eating are dissociable, naloxone infusions into the NAc shell or VP, but not the basolateral amygdala (BLA), decreased the palatability of sucrose solutions (Wassum et al., 2009). However, when the μ-opioid receptor antagonists naloxone or CTOP were infused into the BLA, but not NAc shell or VP, there was a marked attenuation of the increased motivation to respond for sucrose solutions typically seen in a hungry state ( [Wassum et al., 2011] and [Wassum et al., 2009]). This suggests that the incentive properties of sucrose are regulated by amygdalar circuitries. Overall, the above findings show that different aspects of hedonic eating, such as processing of information related to food palatability, approach behaviors, and increases in the incentive value of palatable food in hungry animals, are differentially regulated by discrete microcircuitries within the context of a larger corticolimbic-striatopallidal-hypothalamic-thalamocortical circuitry (Figure 2).
Do Adaptations in Brain Hedonic Circuitries Drive Compulsive Eating?
The functional relevance of hedonic hotspots in the NAc shell and their influence on broader feeding-related circuitries in the brain have been considered by Kelley et al. (2005). They hypothesize that the NAc shell→LH pathway, along with upstream and downstream regulatory brain regions (Figure 2), serve a “sentinel” purpose (Kelley et al., 2005). Specifically, they propose that even in hungry animals when the drive to eat is strong, the ability to cease feeding behavior must be retained in case of threats from the environment (Kelley et al., 2005). As such, activation of NAc shell neurons and concomitant inhibition of LH neurons may disrupt ongoing feeding and facilitate switching of behavior to more appropriate adaptive responses, such as freezing or escape (Kelley et al., 2005). If this is indeed the case, then it will be important to investigate whether this NAc shell→LH control pathway is compromised by overconsumption of palatable food or by genetic factors that influence vulnerability to obesity. With this in mind, our laboratory and others recently reported that overconsumption of palatable calorie-dense food is associated with the emergence of compulsive-like feeding behavior in rats ( [Johnson and Kenny, 2010], [Latagliata et al., 2010] and [Oswald et al., 2010]). Specifically, we found that palatable food consumption in obese rats was resistant to disruption by an aversive conditioned stimulus that predicted negative outcome (electric footshock) (Johnson and Kenny, 2010). Thus, it will be important to test whether deficits in the NAc shell→LH control pathway triggered by overeating at least partially contributes to the conspicuous failure of overweight and obese individuals to utilize information regarding the deleterious consequences of their consummatory behavior to moderate their food intake.
Altered Brain Reward Activity in Obesity: Human Brain Imaging Studies
Obtaining the stimulatory effects of palatable food on brain reward systems is considered an important motivational factor contributing to overeating. Thus, an important question is whether alterations in brain reward function may contribute to the development of obesity. An intuitive prediction is that enhanced constitutive responsiveness of brain reward systems to palatable food would result in overeating and weight gain. Consistent with this hypothesis, individuals with high levels of trait reward sensitivity display enhanced activity in brain regions implicated in food reward, including the NAc, amygdala, OFC, and VP, upon exposure to palatable food like chocolate cake and pizza (Beaver et al., 2006). Obese individuals similarly demonstrate increased activation of brain reward circuitries in response to palatable food or food-associated cues compared with lean controls ( [Gautier et al., 2000], [Karhunen et al., 1997] and [Rothemund et al., 2007]). High levels of trait reward sensitivity were also correlated with increased body weight in human subjects ( [Davis et al., 2004] and [Franken and Muris, 2005]). Importantly, however, obese woman (BMI > 30) had higher levels of anhedonia (i.e., diminished baseline sensitivity to reward) than overweight woman (BMI > 25 < 30) (Davis et al., 2004). Similarly, Stice and coworkers (2008b) have shown that obese adolescent girls had increased activation of the insula and other cortical brain regions in response to palatable food or food-associated cues compared with lean control subjects, but that activation of the caudate area of the striatum in response to the palatable food was inversely correlated with BMI in the obese subjects. Moreover, women who gained weight over a 6 month period had a marked decline in striatal activity in response to palatable food during this time period compared with women who did not gain weight (Stice et al., 2010a). Taking all of this together, it appears that hypersensitivity of reward circuitries may predispose an individual to overeating and weight gain (Stice et al., 2010b). However, as weight gain increases then deficits in the activity of specific components of the brain reward system, particularly the striatum, may begin to emerge. It has been proposed that the emergence of this state of reward hyposensitivty may perpetuate the overconsumption of palatable food in order to overcome such reward deficits ( [Stice et al., 2008a] and [Wang et al., 2002]). Hence, too little or too much food reward seems to increase vulnerability to overeating and obesity (Stoeckel, 2010). An attractive conceptual framework for reconciling these apparently opposing viewpoints is that corticolimbic areas involved in organizing behavior toward obtaining food rewards and making predictions about anticipated future food reward may become hyperactive in overweight individuals and those predisposed to obesity. Conversely, striatal brain sites that process the actual experience of pleasure from hedonic eating may become gradually less functional in these same individuals. The relative motivational value of palatable food would therefore be expected to increase during the development of obesity at the same time that the hedonic value obtained from consuming palatable food decreases. These adaptations would act in a concerted manner to drive the ever-increasing motivation to consume palatable food.
Altered Brain Reward Activity in Obesity: Rodent Studies
The effects of palatable food consumption on brain reward systems have been directly assessed in laboratory animals using the brain-stimulation reward (BSR) procedure. It is well known that electrical stimulation of the LH, which receives tonic inhibitory input from accumbal hedonic hot spots (Figure 2), is highly rewarding and rats will work hard to self-stimulate this brain region, e.g., (Markou and Frank, 1987). In addition to supporting self-stimulation behavior, electrical stimulation of LH can also induce intense bouts of feeding behavior (Margules and Olds, 1962), and it has been proposed that the rewarding properties of LH stimulation may be related to the intrinsic role of this brain site in the appetitive and incentive properties of food (Margules and Olds, 1962). Consistent with this view, hunger and weight loss increase the sensitivity of rats to the rewarding value LH self-stimulation ( [Blundell and Herberg, 1968], [Carr and Simon, 1984] and [Margules and Olds, 1962]), an effect that can be blocked by intracerebroventricular infusion of leptin (Fulton et al., 2000). Conversely, electric self-stimulation of the LH is inhibited in sated animals (Wilkinson and Peele, 1962). Indeed, overfeeding of rats through intragastric feeding tube (Hoebel and Teitelbaum, 1962), gastric distention, or intravenous glucagon infusion that mimics postprandial satiety ( [Hoebel, 1969], [Hoebel and Balagura, 1967] and [Mount and Hoebel, 1967]), all attenuate responding for LH stimulation. In fact, rats which previously responded vigorously for rewarding LH stimulation will respond as if this stimulation were aversive after food intake or development of obesity (Hoebel and Thompson, 1969). Hence, chronic food restriction and weight loss enhances, whereas overfeeding diminishes, the sensitivity of reward-related sites in the LH. The sensitivity of LH neurons to rewarding electrical self-stimulation may therefore provide important insights into the functioning of brain circuitry that regulates hedonic responses to food.
As ease of access to energy-dense palatable food and consequent overconsumption is considered a major environmental factor contributing to obesity (Volkow and Wise, 2005), our laboratory recently utilized the BSR procedure to assess brain reward activity in rats with extended access to palatable food. Specifically, we recorded responding for electrical stimulation of the LH in rats that had ad libitum access to nutritional chow alone or chow in combination with 18–23 hr daily access to a palatable diet. This diet consisted of cheesecake, bacon, sausage, and other appetizing food items (Johnson and Kenny, 2010). We found that rats with extended access to the palatable food rapidly gained significant amounts of weight and demonstrated a progressively worsening brain reward deficit (reflected as diminished responsiveness to rewarding LH stimulation) (Johnson and Kenny, 2010; Figure 3). This suggests that development of diet-induced obesity is associated with a gradual decrease in the responsiveness of reward sites in the LH (Johnson and Kenny, 2010). Deficits in reward signaling have also been reported in adult rats that previously had unlimited access to sucrose or high-fat food during adolescence ( [Teegarden et al., 2009], [Vendruscolo et al., 2010a] and [Vendruscolo et al., 2010b]). These effects in rats are reminiscent of the decreased striatal activation in response to food reward described above in human subjects as they gained weight over a 6 month period (Stice et al., 2010a; see Figure 4). Such diet-induced reward deficits in overweight rats, and perhaps in humans that gain weight, likely reflect a counteradaptive response in food reward circuitries to oppose their overstimulation by palatable food (Johnson and Kenny, 2010). An important aspect of this finding is that similar deficits in reward function are also detected in rats that overconsume cocaine or heroin ( [Ahmed et al., 2002], [Kenny et al., 2006] and [Markou and Koob, 1991]; Figure 3). In fact, it has been hypothesized that drug-induced reward dysfunction may contribute to the transition from controlled to uncontrolled drug use by providing a new source of motivation to consume the drug in order to alleviate the persistent state of diminished reward ( [Ahmed and Koob, 2005] and [Koob and Le Moal, 2008]). Therefore, it is possible that deficits in the sensitivity of reward sites in the LH induced by overeating may increase the long-term persistence of palatable food consumption in overweight rats by shifting dietary preference toward food with higher hedonic impact to alleviate the persistent state of negative reward.
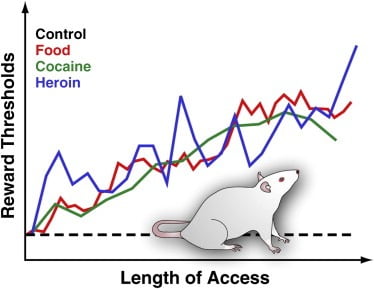
Figure 3. Reward Thresholds in Rats with Extended Daily Access to Palatable Food, Cocaine, or Heroin
To measure reward thresholds, a stimulating electrode is surgically implanted into the lateral hypothalamus of rats, a region in which electrical stimulation is powerfully rewarding and can trigger intense bouts of feeding behavior. After recovery, animals are allowed to self-stimulate this region by turning a wheel. After stable self-stimulation behavior is established, the minimum stimulation intensity that maintained self-stimulation behavior is determined (i.e., the reward threshold). This reward threshold provides an operational measure of the activity of the reward system. Reward thresholds remain stable and unaltered in control rats that have access to standard lab chow and that remain drug naïve. However, thresholds gradually elevate in rats with extended daily access to an energy-dense palatable diet consisting of tasty food items (e.g., cheesecake, bacon, chocolate, etc.). Similarly, reward thresholds progressively elevated in rats that have extended daily access to intravenous cocaine or heroin infusions. Elevated rewards threshold are interpreted to reflect decreased sensitivity of the brain reward system. These effects suggest that overconsumption of palatable food and associated weight gain can induce profound deficits in brain reward similar to those induced by excessive consumption of addictive drug
Figure 4. Striatal Plasticity in ObesityWeight gain is associated with decreased striatal activation in response to palatable food, as measured by fMRI, and lower levels of striatal dopamine D2 receptor (D2R) availability in humans (see text for details).
Deficient Dopamine D2 Receptor Signaling in Obesity
Several recent reports have revealed potential mechanisms through which reward deficits may emerge in response to overconsumption of palatable food during the development of obesity. As noted above, women who gained weight over a 6 month period had a marked decline in striatal activity in response to palatable food over this time period compared with women who did not gain weight (Stice et al., 2010a; Figure 4). Fasted individuals permitted to eat their favorite meal to satiety had lower levels of binding of the dopamine D2 receptor (D2R) antagonist raclopride in striatum (Small et al., 2003), suggesting that D2R signaling decreases in response to palatable food consumption. Indeed, obese individuals have lower levels of striatal D2R availability compared with lean controls ( [Barnard et al., 2009], [Stice et al., 2008a] and [Wang et al., 2001]; Figure 4), whereas weight loss in obese patients is associated with increased striatal D2R density (Wang et al., 2008). Considering that striatal dopamine transmission plays a key role in regulating hedonic eating, adaptive decreases in D2R signaling could contribute to the reduced responsiveness of the striatum to palatable food in obese individuals. To test this possibility, Small and coworkers examined activity in brain reward circuitries in response to a palatable milkshake in control individuals and those carrying the TaqIA A1 allele (Felsted et al., 2010). The TaqIA restriction fragment length polymorphism is downstream from the D2R gene (Neville et al., 2004), and individuals carrying the A1 allele of the polymorphism have between 30%–40% fewer striatal D2Rs compared with those not carrying the allele ( [Jönsson et al., 1999], [Ritchie and Noble, 2003] and [Stice et al., 2010b]). In addition, A1 allele carriers also have reduced glucose metabolism in striatal and cortical brain areas involved in hedonic responses to food (Jönsson et al., 1999). Individuals harboring the TaqIA A1 allele are overrepresented in obese populations ( [Barnard et al., 2009], [Stice et al., 2008a] and [Wang et al., 2001]). Furthermore, the A1 allele also increases vulnerability to alcohol, opioid, and psychomotor stimulant addiction ( [Lawford et al., 2000], [Noble et al., 1993] and [Noble et al., 2000]). It was found that midbrain areas likely including the VTA and SN, which provide dopaminergic input to the striatum, were activated in response to a palatable milkshake in control individuals (Felsted et al., 2010). Conversely, activity in these brain sites was actually decreased in response to food reward in the A1 allele carriers (Felsted et al., 2010). Similar inverse responses in brain activation between A1 allelic carriers and noncarriers were also detected in thalamic and cortical brain sites (Felsted et al., 2010). These data are highly consistent with a key role for D2Rs in regulating mesostriatal responsiveness to palatable food. Stice and colleagues (2008a) found an inverse correlation between BMI and activation of the striatum (caudate and putamen) in response to palatable chocolate milkshake in human patients. Moreover, this inverse relationship was most apparent in individuals carrying the TaqIA A1 allele (Stice et al., 2008a). Future weight gain in these individuals, measured 1 year after initial brain imaging, showed that the magnitude of striatal activation in response to palatable food was negatively correlated with weight gain in subjects with the A1 allele and positively correlated in the non-A1 allele subjects (Stice et al., 2008a). In a follow-up study, it was reported that the magnitude of striatal activation in response to imagined eating of palatable food, as opposed to its actual consumption, was inversely correlated with weight gain over the following year in subjects with the A1 allele but positively correlated in non-A1 allele subjects (Stice et al., 2010b). These findings suggest that D2Rs regulate striatal responsiveness to palatable food and that diminished D2R signaling induced by weight gain or genetic factors may increase vulnerability to obesity. Hence, common genetic vulnerabilities to reward dysfunction upon overconsumption of palatable food or drugs of abuse may increase predisposition to obesity and drug addiction, respectively.
Deficient D2R Signaling Contributes to Reward Deficits in Obesity
Similar to the downregulated striatal D2R levels in obese human subjects, D2R levels are also lowered in mice and rats fed a palatable diet (e.g., [Colantuoni et al., 2001], [Geiger et al., 2009] and [Johnson and Kenny, 2010]) and in rats genetically predisposed to obesity (Zucker rats) (Thanos et al., 2008). Our laboratory has directly investigated the role for disrupted striatal dopamine transmission in general, and decreases in D2R signaling in particular, in the addiction-like reward deficits that emerge in rats during the development of obesity (see Figure 5). Specifically, we tested the effects of decreasing the expression of striatal D2Rs in rats using viral-mediated RNA interference, then assessing BSR thresholds when rats had access to chow only or chow in combination with 18–23 hr daily access to a palatable high-energy diet (i.e., a cafeteria diet) (Johnson and Kenny, 2010). We found that responding for rewarding LH stimulation began to decrease almost immediately upon exposure to the cafeteria diet in the D2R knockdown rats (Johnson and Kenny, 2010; Figure 5). Decreases in striatal D2R levels therefore rapidly accelerate the emergence of reward hypofunctionality in rats with extended access to highly palatable food, a process that typically takes many weeks to emerge in control rats with extended access to palatable diet. However, knockdown of striatal D2Rs in rats with access to chow only did not alter responding for rewarding LH stimulation, suggesting that diminished striatal D2R signaling interacts with other diet-induced adaptive responses in brain reward circuitries to trigger reward hyposensitivity. In addition to lowered D2R levels, other aspects of striatal dopaminergic transmission are also altered in the brains of obese rats. For example, Sprague-Dawley rats bred selectivity to rapidly gain weight on a high-energy diet (obesity prone rats) have lower basal and evoked dopamine levels in the NAc than rats that are resistant to weight gain (obesity resistant rats) (Geiger et al., 2008; see also Rada et al., 2010). The obesity prone rats also have decreased levels of dopamine biosynthetic and storage machinery, suggesting that a failure in the production and release of dopamine contributes to deficits in striatum dopamine transmission in obese rats (Geiger et al., 2008). Rats that developed obesity through overconsumption of a palatable high-energy diet also had lower basal and evoked dopamine levels in the NAc compared with rats that had access only to standard chow ( [Davis et al., 2008] and [Geiger et al., 2009]). Importantly, a meal of standard chow was sufficient to increase dopamine levels in the NAc of the control rats, whereas only the highly palatable food items were sufficient to trigger accumbal dopamine release in the obese rats that had a history of extended access to the palatable food (Geiger et al., 2009). These findings demonstrate that the development of obesity in rats is associated with dysfunction in mesostriatal dopamine transmission, most prominently at striatal D2Rs, and that deficient D2R signaling contributes to the emergence of reward deficits during the development of obesity in rats. This is consistent with the fact that downregulation of striatal D2Rs is a notable neuroadaptive response to weight gain in humans ( [Barnard et al., 2009], [Stice et al., 2008a] and [Wang et al., 2001]), and that deficient striatal D2R signaling may blunt striatal responses to hedonic food in human subjects, thereby predisposing the individual to future weight gain ( [Stice et al., 2008a] and [Wang et al., 2001]). It has therefore been hypothesized that the persistent overconsumption of palatable food may represent an attempt to alleviate deficient striatal D2R signaling and associated reward deficits in obese individuals ( [Geiger et al., 2009], [Johnson and Kenny, 2010], [Stice et al., 2008a] and [Wang et al., 2002]).
Figure 5. D2 Dopamine Receptors, Reward Dysfunction, and Compulsivity in Obesity – Knockdown of dopamine D2 receptors (D2R) knockdown in rat striatum accelerates the emergence of reward dysfunction and compulsive eating in rats with extended access to palatable food.
Deficient D2R Signaling May Trigger Compulsive Eating in Obesity
Obesity is characterized by overeating that persists despite an expressed desire to limit consumption and knowledge of the profound negative health and social consequences of continued excessive consumption ( [Booth et al., 2008], [Delin et al., 1997] and [Puhl et al., 2008]). This is exemplified by the fact that many obese patients will undergo potentially dangerous bariatric (gastric bypass) surgery to control their weight (Yurcisin et al., 2009), yet often relapse to overeating even though the surgery decreases subjective ratings of hunger and reduces the capacity to consume large quantities of food ( [Kalarchian et al., 2002] and [Saunders, 2001]). Drug addiction is similarly defined as a loss of inhibitory control over drug consumption and persistence in the habit despite an awareness of the potentially devastating health, social, or financial consequences (DSM-IV; American Psychiatric Association, 1994). As such, obesity and drug addiction share the hallmarks of compulsive disorders in that there is a conspicuous failure to utilize information regarding future deleterious consequences to moderate consumption and persistence in consumption despite availability of less harmful alternative behaviors.
Compulsive drug taking has been operationally defined in rodents as consumption that is resistant to suppression by punishment or environmental stimuli predicting punishment ( [Pelloux et al., 2007] and [Vanderschuren and Everitt, 2004]). Periods of extended access to cocaine and other drugs of abuse can drive the emergence of compulsive drug-taking behaviors in rats ( [Ahmed and Koob, 1998], [Deroche-Gamonet et al., 2004] and [Vanderschuren and Everitt, 2004]). Indeed, rats with a history of extensive cocaine consumption display intake that is resistant to disruption by an aversive conditioned stimulus (CS) predicting negative outcome (i.e., a cue light that predicts delivery of aversive footshock) ( [Belin et al., 2008] and [Vanderschuren and Everitt, 2004]). Conversely, the same aversive CS can profoundly decrease drug-seeking responses in rats with relatively limited access to the drug. Considering the similarities between compulsive drug use in addiction and overeating in obesity, we recently investigated whether obese rats would consume palatable food in a compulsive-liked manner and if striatal D2Rs play a role in this process (Johnson and Kenny, 2010). We found that obese rats with a history of extended access to palatable food continued to eat palatable food even in the presence of a noxious CS (a light cue) that predicted the delivery of aversive footshock (Johnson and Kenny, 2010). In contrast, the same aversive CS disrupted palatable food consumption in lean rats with very limited exposure to energy-dense palatable food. Palatable food consumption can therefore become compulsive in obese rats in much the same way that cocaine consumption can become compulsive. Consistent with this interpretation of the data, mice that previously had access to a palatable high-fat diet spent more time in an aversive environment (brightly lit) to obtain the palatable food than mice that had no prior experience of the diet (Teegarden and Bale, 2007). Because of fear of predation, brightly lit open arenas are highly aversive to mice (Suarez and Gallup, 1981). Mice therefore become resistant to the potentially negative consequences of their foraging behavior and will risk predation to obtain palatable food even when less palatable food is available at a far lower peril.
Intriguingly, the A1 allele of the TaqIA polymorphism that results in lowered striatal D2R density (Noble, 2000) and blunted striatal activation in response to palatable food (Stice et al., 2008a) is also associated with deficits in learning to avoid actions that have negative consequences (Klein et al., 2007). It is precisely this type of failure to utilize information related to future negative consequences of overeating that may contribute to the development of compulsive eating in obese individuals. We found that the emergence of compulsive-like eating in rats with access to palatable food was dramatically accelerated following striatal D2R knockdown (Johnson and Kenny, 2010). In fact, rats with striatal D2R knockdown that previously had only 14 days of extended access to energy-dense palatable food displayed palatable food consumption that was resistant to disruption by an aversive CS (Johnson and Kenny, 2010; Figure 5). However, this 14 day period of limited exposure to the palatable food was not sufficient to induce compulsive-like eating in control rats (Johnson and Kenny, 2010). These findings suggest that addiction-like compulsive intake of palatable food can emerge in obese rats. Furthermore, deficient striatal D2R signaling, which accelerates the emergence of reward hyposensitivity in response to palatable food overconsumption, also accelerates the emergence of compulsive-like eating (Figure 5).
Interactions between D2Rs and Hormonal Regulators of Energy Balance in Obesity
Exogenous leptin administered into the VTA inhibits mesoaccumbens dopamine transmission and feeding behavior ( [Hommel et al., 2006] and [Krügel et al., 2003]). In addition to its acute inhibitory effect on midbrain dopamine systems, there is accumulating evidence that tonic leptin signaling may also be necessary to maintain appropriate levels of mesostriatal dopamine signaling. Flier and colleagues found that ob/ob mice had lower levels of tyrosine hydroxylase in midbrain dopamine neurons, a key enzyme in the biosynthesis of dopamine (Fulton et al., 2006). In addition, ob/ob mice had reduced evoked dopamine release into the NAc (Fulton et al., 2006) and decreased somatodendritic vesicular stores of dopamine in VTA and SN (Roseberry et al., 2007). These deficiencies in dopamine production and signal transduction machinery in ob/ob mice were corrected by treatment with leptin ( [Fulton et al., 2006] and [Pfaffly et al., 2010]). In fact, leptin infused only into the LH was sufficient to correct dysfunctional dopamine transmission in ob/ob mice (Leinninger et al., 2009), suggesting the leptin receptor-expressing cells in the LH act to maintain appropriate levels of dopamine signaling. In addition to diminished production and release of dopamine, ob/ob mice also tended to have lower levels of D2R expression in striatum (Pfaffly et al., 2010). Furthermore, exogenous leptin treatment under a regimen that results in the development of insensitivity to leptin signaling (i.e., leptin resistance) markedly lowered striatal D2R levels in wild-type mice (Pfaffly et al., 2010). Obese rats develop leptin resistance in the VTA ( [Matheny et al., 2011] and [Scarpace et al., 2010]) and also have lower levels of TH in VTA, diminished dopamine release in striatum, and reduced striatal D2R levels (Geiger et al., 2008). Taken together, these data demonstrate that leptin has complex actions on midbrain dopamine systems. On the one hand, acute activation of leptin receptors in the VTA exerts an inhibitory effect on mesoaccumbens dopamine transmission and can inhibit feeding behavior ( [Hommel et al., 2006] and [Krügel et al., 2003]). On the other, leptin signaling in the midbrain is necessary to maintain appropriate dopamine production and signal transmission, and genetic deficits in leptin signaling or the development of leptin resistance in obesity profoundly disrupts mesoaccumbens dopamine systems. As such, it is an interesting possibility that the development of leptin resistance in midbrain dopaminergic neurons during the development of obesity may play a central role in the disruption of striatal D2R signaling that appears to drive the emergence of addiction-like reward dysfunction and compulsive overeating (Figure 5) in obese rats.
In addition to the complex effects of leptin signaling on mesostriatal dopaminergic transmission, there is accumulating evidence that D2Rs may in turn regulate leptin signaling. The D2R agonist bromocriptine reduces circulating levels of leptin ( [Doknic et al., 2002], [Kok et al., 2006] and [Mastronardi et al., 2001]), suggesting that D2Rs exert an inhibitory influence on leptin levels. Furthermore, mice with null mutation in the D2R gene have enhanced leptin signaling in hypothalamus and are more sensitive to the anorectic effects of leptin (Kim et al., 2010). It is well known that leptin levels increase during the development of obesity (hyperleptinemia), yet there is a concomitant decrease in sensitivity to leptin signaling (i.e., leptin resistance) (Hamilton et al., 1995). Thus, it is an interesting possibility that decreases in striatal D2R signaling during the development of obesity may represent a compensatory response to overconsumption of palatable food and weight gain that increases circulating leptin levels and increases its signaling efficiency to overcome the development of leptin resistance. Such an action may enhance the inhibitory effects of leptin on striatal systems that regulate responsiveness to palatable food ( [Farooqi et al., 2007], [Fulton et al., 2006] and [Hommel et al., 2006]), thereby acting to attenuate hedonic responses to palatable food. Putting this finding together with the regulatory role of leptin on D2Rs described above, it seems that leptin and D2R signaling may be coupled in a reciprocal manner to regulate homeostatic and hedonic aspects of feeding behavior.
Dysregulated Brain Stress Systems in Obesity
Leptin signaling in the midbrain acts to regulate mesoaccumbens dopamine transmission and responsiveness to hedonic food. However, neurons in the VTA that express leptin receptors project sparsely to the NAc, and instead demonstrate more prominent projections to the central nucleus of the amygdala (CeA) (Leshan et al., 2010). In the context of feeding behavior, the CeA is known to regulate the inhibitory effects of noxious environmental stimuli on food consumption (Petrovich et al., 2009). Specifically, lesions of the CeA, but not basolateral amygdala (BLA), abolish the inhibitory effects of a footshock-paired conditioned stimulus on feeding, suggesting that the CeA is critical for inhibitory control over feeding behavior in response to environmental cues predicting negative outcome (Petrovich et al., 2009). Obese rats, or nonobese rats with striatal D2R knockdown and access to palatable food, continue to consume palatable food in a compulsive-like manner in the presence of an aversive CS. These effects are strikingly similar to the disruption in reactivity to an aversive CS in CeA lesioned rats (Petrovich et al., 2009). Thus, it will be important to determine if alterations in CeA activity, perhaps as a consequence of striatal D2R downregulation or development of leptin resistance in midbrain, contributes to the emergence of compulsive-like eating in obese rats.
In addition to regulating the effects of noxious environmental stimuli on feeding behavior, the CeA may also play a key role in stress-related hedonic eating. In humans, stress powerfully motivates the selection and consumption of energy-dense palatable food independent of caloric need ( [Gluck et al., 2004], [Kandiah et al., 2006] and [O’Connor et al., 2008]), and obesity is associated with elevated stress-related glucocorticoid secretion ( [Björntorp and Rosmond, 2000] and [la Fleur, 2006]). Environmental and social stressors also induce hyperphagia of energy-dense palatable food in rodents and monkeys, with palatable food consumption potentially attenuating adverse effects of stress ( [Dallman et al., 2003], [Dallman et al., 2006], [Pecoraro et al., 2004] and [Wilson et al., 2008]). Further, the stress-evoking drug yohimbine can reinstate previously extinguished palatable food seeking responses (lever-pressing) in rats, an effect attenuated by the corticotropin-releasing factor-1 (CRF-1) receptor antagonist antalarmin (Ghitza et al., 2006). It is important to point out that the precise effects of stress on food consumption in humans and laboratory animals is dependent on the nature and magnitude of the stressor, the type of food available for consumption (palatable versus bland), body weight and gender (Dallman, 2010).
Mice with extended access to a palatable high-fat diet had decreased expression of the stress hormone CRF in the CeA (Teegarden and Bale, 2007). Conversely, mice undergoing “withdrawal” from the palatable diet had increased CRF expression in the CeA (Teegarden and Bale, 2007). This latter effect is very similar to the increased CRF expression in CeA detected in rats undergoing withdrawal from all major drugs of abuse (Koob, 2010). Because further drug use can normalize this aversive neurobiological response to drug withdrawal, it has been hypothesized that hyperactive CRF transmission in CeA and other limbic structures may facilitate the development of compulsive drug use (Koob and Zorrilla, 2010). Consistent with this view, mice undergoing withdrawal from palatable energy-dense food, which had elevated CRF levels in CeA, spent significantly more time in an aversive (brightly lit) environment to obtain palatable food than mice with no prior experience of the food, even though less palatable was available in a nonaversive (darkly lit) environment (Teegarden and Bale, 2007). Mice therefore become resistant to the potentially negative consequences of their foraging behavior and risked predation to obtain palatable food even when less palatable food is available at a far lower peril, in part to attenuate CRF transmission in the CeA (Teegarden and Bale, 2007). Several further pieces of evidence support a role for CRF transmission in compulsive eating. In particular, a recent study assessed the effects of the CRF-1 receptor antagonist R121919 on food consumption in rats undergoing cyclic variation in their diet in which they had access to standard chow 5 days per week and access to a palatable sugar diet 2 days per week (Cottone et al., 2009). After 7 weeks of this cyclic variation in diet, R121919 attenuated the excessive consumption of the highly palatable diet and increased consumption of bland chow (Cottone et al., 2009). Furthermore, CRF expression levels in the CeA were increased in the cycled rats during withdrawal from the palatable diet but returned to baseline levels after re-exposure to the palatable diet (Cottone et al., 2009). Finally, CRF regulation of GABAergic transmission in the CeA was enhanced in the cycled rats undergoing withdrawal from the palatable diet compared with control rats that previously had access only to bland chow, reflected in the fact that R121919 disrupted evoked GABAergic inhibitory post-synaptic potential in CeA slices from the cycled rats at a concentration that did not alter transmission in slices from the control rats (Cottone et al., 2009). Interestingly, a similar effect of CRF on GABAergic transmission in CeA has also been observed in rats undergoing withdrawal from chronic ethanol exposure (Roberto et al., 2010). Finally, the CeA, bed nucleus of the stria terminalis (BNST), and NAc shell constitute a larger contiguous structure termed the “extended amygdala.” Infusion of CRF into the NAc shell at sites that stimulates hedonic eating enhances the motivational salience of an environmental cue that had previously been paired with the availability of palatable food (Peciña et al., 2006a). Stress may therefore enhance the salience of food-paired environmental cues by modulating the activity of NAc shell neurons. Taken together, these findings suggest that excessive consumption palatable food or drugs of abuse may induce similar neuroadaptive responses in extrahypothalamic stress pathways in the brain, which may contribute to compulsive consummatory behaviors. It will be interesting, therefore, to assess the role extrahypothalamic stress pathways in regulating the deficits in striatal D2R signaling, reward hypofunctionality, and compulsive-like feeding behaviors that emerge in rats during the development of obesity.
Conclusions
Much progress has been made recently in identifying brain systems involved in hedonic effects of palatable food and the adaptations that occur in these circuitries in response to overconsumption of palatable food and weight gain. It is striking that similar brain systems and common adaptive responses are triggered in response to consumption of both palatable food and addictive drugs. In particular, overconsumption of palatable food or drugs of abuse induces similar deficits in the responsiveness of brain reward systems. Palatable food and addictive drugs induce deficits in striatal dopamine transmission and the expression of striatal D2Rs. Palatable food and addictive drugs also triggers the emergence of compulsive-like consummatory behavior in rodents and engage extrahypothalamic stress responses. Finally, common genetic vulnerabilities in brain reward systems may predispose individuals to overeating and obesity and also drug use and addiction. In fact, based on these and other similarities between obesity and drug addiction, it has been argued that obesity should be included as a diagnostic category in the forthcoming DSM-V ( [Devlin, 2007] and [Volkow and O’Brien, 2007]). With this in mind, critical areas for future research will involve further investigating the potential neurobiological overlaps between compulsive overeating and drug use. For example, it will be important to test whether obesity is related to the development of habit-like consummatory behavior resulting from plasticity in dorsal striatum in the same way that drug addiction may be related to striatal remodeling and the emergence of habit-like drug seeking behaviors ( [Everitt and Robbins, 2005], [Hollander et al., 2010] and [Kasanetz et al., 2010]). Also, cortical brain regions involved in executive control and decision making (i.e., prefrontal cortex) and in interoceptive processing (insular cortex) have been heavily implicated in drug addiction ( [Everitt et al., 2008], [Fineberg et al., 2010], [Koob and Volkow, 2010] and [Naqvi and Bechara, 2009]), yet little is known of their precise role in compulsive eating and obesity, e.g., ( [Nair et al., 2011] and [Volkow et al., 2009]). Taken together, the data reviewed above support the notion that obesity and drug addiction may arise from similar neuroadaptive responses in brain reward circuitries, and suggest that known mechanisms of addiction may provide a heuristic framework for understanding obesity.
Acknowledgments
The author is supported by grants from the National Institute on Drug Abuse (NIDA). The author is grateful to Paul Johnson and Christie Fowler for valuable insights and comments on the manuscript. This is manuscript number 21042 from The Scripps Research Institute.
References
1.
o Abizaid et al., 2006a
o A. Abizaid, Q. Gao, T.L. Horvath
o Thoughts for food: Brain mechanisms and peripheral energy balance
o Neuron, 51 (2006), pp. 691–702
o
2.
o Abizaid et al., 2006b
o A. Abizaid, Z.W. Liu, Z.B. Andrews, M. Shanabrough, E. Borok, J.D. Elsworth, R.H. Roth, M.W. Sleeman, M.R. Picciotto, M.H. Tschöp et al.
o Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite
o J. Clin. Invest., 116 (2006), pp. 3229–3239
o
3.
o Ahmed and Koob, 1998
o S.H. Ahmed, G.F. Koob
o Transition from moderate to excessive drug intake: Change in hedonic set point
o Science, 282 (1998), pp. 298–300
o
4.
o Ahmed and Koob, 2005
o S.H. Ahmed, G.F. Koob
o Transition to drug addiction: A negative reinforcement model based on an allostatic decrease in reward function
o Psychopharmacology (Berl.), 180 (2005), pp. 473–490
o
5.
o Ahmed et al., 2002
o S.H. Ahmed, P.J. Kenny, G.F. Koob, A. Markou
o Neurobiological evidence for hedonic allostasis associated with escalating cocaine use
o Nat. Neurosci., 5 (2002), pp. 625–626
o
6.
o Allison et al., 1999
o D.B. Allison, K.R. Fontaine, J.E. Manson, J. Stevens, T.B. VanItallie
o Annual deaths attributable to obesity in the United States
o JAMA, 282 (1999), pp. 1530–1538
o
7.
o American Psychiatric Association, 1994
o American Psychiatric Association
o Diagnostic and Statistical Manual of Mental Disorders
o (Fourth Edition)American Psychiatric Association, Washington, DC (1994)
o
8.
o Angeles-Castellanos et al., 2007
o M. Angeles-Castellanos, J. Mendoza, C. Escobar
o Restricted feeding schedules phase shift daily rhythms of c-Fos and protein Per1 immunoreactivity in corticolimbic regions in rats
o Neuroscience, 144 (2007), pp. 344–355
o
9.
o Baicy et al., 2007
o K. Baicy, E.D. London, J. Monterosso, M.L. Wong, T. Delibasi, A. Sharma, J. Licinio
o Leptin replacement alters brain response to food cues in genetically leptin-deficient adults
o Proc. Natl. Acad. Sci. USA, 104 (2007), pp. 18276–18279
o
10.
o Baldo et al., 2004
o B.A. Baldo, L. Gual-Bonilla, K. Sijapati, R.A. Daniel, C.F. Landry, A.E. Kelley
o Activation of a subpopulation of orexin/hypocretin-containing hypothalamic neurons by GABAA receptor-mediated inhibition of the nucleus accumbens shell, but not by exposure to a novel environment
o Eur. J. Neurosci., 19 (2004), pp. 376–386
o
11.
o Balleine and Dickinson, 2000
o B.W. Balleine, A. Dickinson
o The effect of lesions of the insular cortex on instrumental conditioning: evidence for a role in incentive memory
o J. Neurosci., 20 (2000), pp. 8954–8964
o
12.
o Barnard et al., 2009
o N.D. Barnard, E.P. Noble, T. Ritchie, J. Cohen, D.J. Jenkins, G. Turner-McGrievy, L. Gloede, A.A. Green, H. Ferdowsian
o D2 dopamine receptor Taq1A polymorphism, body weight, and dietary intake in type 2 diabetes
o Nutrition, 25 (2009), pp. 58–65
o
13.
o Basso and Kelley, 1999
o A.M. Basso, A.E. Kelley
o Feeding induced by GABA(A) receptor stimulation within the nucleus accumbens shell: Regional mapping and characterization of macronutrient and taste preference
o Behav. Neurosci., 113 (1999), pp. 324–336
o
14.
o Batterham et al., 2007
o R.L. Batterham, D.H. ffytche, J.M. Rosenthal, F.O. Zelaya, G.J. Barker, D.J. Withers, S.C. Williams
o PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans
o Nature, 450 (2007), pp. 106–109
o
15.
o Baxter and Murray, 2002
o M.G. Baxter, E.A. Murray
o The amygdala and reward
o Nat. Rev. Neurosci., 3 (2002), pp. 563–573
o
16.
o Bean et al., 2008
o M.K. Bean, K. Stewart, M.E. Olbrisch
o Obesity in America: Implications for clinical and health psychologists
o J. Clin. Psychol. Med. Settings, 15 (2008), pp. 214–224
o
17.
o Beaver et al., 2006
o J.D. Beaver, A.D. Lawrence, J. van Ditzhuijzen, M.H. Davis, A. Woods, A.J. Calder
o Individual differences in reward drive predict neural responses to images of food
o J. Neurosci., 26 (2006), pp. 5160–5166
o
18.
o Belin et al., 2008
o D. Belin, A.C. Mar, J.W. Dalley, T.W. Robbins, B.J. Everitt
o High impulsivity predicts the switch to compulsive cocaine-taking
o Science, 320 (2008), pp. 1352–1355
o
19.
o Berridge, 1996
o K.C. Berridge
o Food reward: Brain substrates of wanting and liking
o Neurosci. Biobehav. Rev., 20 (1996), pp. 1–25
o
20.
o Berridge, 2009
o K.C. Berridge
o ‘Liking’ and ‘wanting’ food rewards: Brain substrates and roles in eating disorders
o Physiol. Behav., 97 (2009), pp. 537–550
o
21.
o Björntorp and Rosmond, 2000
o P. Björntorp, R. Rosmond
o Obesity and cortisol
o Nutrition, 16 (2000), pp. 924–936
o
22.
o Blundell and Herberg, 1968
o J.E. Blundell, L.J. Herberg
o Relative effects of nutritional deficit and deprivation period on rate of electrical self-stimulation of lateral hypothalamus
o Nature, 219 (1968), pp. 627–628
o
23.
o Booth et al., 2008
o M.L. Booth, R.L. Wilkenfeld, D.L. Pagnini, S.L. Booth, L.A. King
o Perceptions of adolescents on overweight and obesity: The weight of opinion study
o J. Paediatr. Child Health, 44 (2008), pp. 248–252
o
24.
o Bragulat et al., 2010
o V. Bragulat, M. Dzemidzic, C. Bruno, C.A. Cox, T. Talavage, R.V. Considine, D.A. Kareken
o Food-Related Odor Probes of Brain Reward Circuits during Hunger: A Pilot fMRI Study
o Obesity, Silver Spring, MD (2010)
o
25.
o Cabanac and Johnson, 1983
o M. Cabanac, K.G. Johnson
o Analysis of a conflict between palatability and cold exposure in rats
o Physiol. Behav., 31 (1983), pp. 249–253
o
26.
o Campfield et al., 1995
o L.A. Campfield, F.J. Smith, Y. Guisez, R. Devos, P. Burn
o Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks
o Science, 269 (1995), pp. 546–549
o
27.
o Cannon and Palmiter, 2003
o C.M. Cannon, R.D. Palmiter
o Reward without dopamine
o J. Neurosci., 23 (2003), pp. 10827–10831
o
28.
o Carr and Simon, 1984
o K.D. Carr, E.J. Simon
o Potentiation of reward by hunger is opioid mediated
o Brain Res., 297 (1984), pp. 369–373
o
29.
o Centers for Disease Control and Prevention, 2009
o Centers for Disease Control and Prevention (2009). U.S. Obesity Trends (Atlanta: Centers for Disease Control and Prevention).
o
30.
o Colantuoni et al., 2001
o C. Colantuoni, J. Schwenker, J. McCarthy, P. Rada, B. Ladenheim, J.L. Cadet, G.J. Schwartz, T.H. Moran, B.G. Hoebel
o Excessive sugar intake alters binding to dopamine and mu-opioid receptors in the brain
o Neuroreport, 12 (2001), pp. 3549–3552
o
31.
o Cornelius et al., 2010
o J.R. Cornelius, M. Tippmann-Peikert, N.L. Slocumb, C.F. Frerichs, M.H. Silber
o Impulse control disorders with the use of dopaminergic agents in restless legs syndrome: A case-control study
o Sleep, 33 (2010), pp. 81–87
o
32.
o Cornier et al., 2009
o M.A. Cornier, A.K. Salzberg, D.C. Endly, D.H. Bessesen, D.C. Rojas, J.R. Tregellas
o The effects of overfeeding on the neuronal response to visual food cues in thin and reduced-obese individuals
o PLoS ONE, 4 (2009), p. e6310 http://dx.doi.org/10.1371/journal.pone.0006310
o
33.
o Cottone et al., 2009
o P. Cottone, V. Sabino, M. Roberto, M. Bajo, L. Pockros, J.B. Frihauf, E.M. Fekete, L. Steardo, K.C. Rice, D.E. Grigoriadis et al.
o CRF system recruitment mediates dark side of compulsive eating
o Proc. Natl. Acad. Sci. USA, 106 (2009), pp. 20016–20020
o
34.
o Dagher, 2009
o A. Dagher
o The neurobiology of appetite: Hunger as addiction
o Int. J. Obes. (Lond.), 33 (Suppl 2) (2009), pp. S30–S33
o
35.
o Dagher and Robbins, 2009
o A. Dagher, T.W. Robbins
o Personality, addiction, dopamine: Insights from Parkinson’s disease
o Neuron, 61 (2009), pp. 502–510
o
36.
o Dallman, 2010
o M.F. Dallman
o Stress-induced obesity and the emotional nervous system
o Trends Endocrinol. Metab., 21 (2010), pp. 159–165
o
37.
o Dallman et al., 2003
o M.F. Dallman, N. Pecoraro, S.F. Akana, S.E. La Fleur, F. Gomez, H. Houshyar, M.E. Bell, S. Bhatnagar, K.D. Laugero, S. Manalo
o Chronic stress and obesity: A new view of “comfort food”
o Proc. Natl. Acad. Sci. USA, 100 (2003), pp. 11696–11701
o
38.
o Dallman et al., 2006
o M.F. Dallman, N.C. Pecoraro, S.E. La Fleur, J.P. Warne, A.B. Ginsberg, S.F. Akana, K.C. Laugero, H. Houshyar, A.M. Strack, S. Bhatnagar, M.E. Bell
o Glucocorticoids, chronic stress, and obesity
o Prog. Brain Res., 153 (2006), pp. 75–105
o
39.
o Davis et al., 2004
o C. Davis, S. Strachan, M. Berkson
o Sensitivity to reward: Implications for overeating and overweight
o Appetite, 42 (2004), pp. 131–138
o
40.
o Davis et al., 2008
o J.F. Davis, A.L. Tracy, J.D. Schurdak, M.H. Tschöp, J.W. Lipton, D.J. Clegg, S.C. Benoit
o Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat
o Behav. Neurosci., 122 (2008), pp. 1257–1263
o
41.
o de Araujo et al., 2010
o I.E. de Araujo, X. Ren, J.G. Ferreira
o Metabolic sensing in brain dopamine systems
o Results Probl. Cell Differ., 52 (2010), pp. 69–86
o
42.
o Delin et al., 1997
o C.R. Delin, J.M. Watts, J.L. Saebel, P.G. Anderson
o Eating behavior and the experience of hunger following gastric bypass surgery for morbid obesity
o Obes. Surg., 7 (1997), pp. 405–413
o
43.
o Deroche-Gamonet et al., 2004
o V. Deroche-Gamonet, D. Belin, P.V. Piazza
o Evidence for addiction-like behavior in the rat
o Science, 305 (2004), pp. 1014–1017
o
44.
o Devlin, 2007
o M.J. Devlin
o Is there a place for obesity in DSM-V?
o Int. J. Eat. Disord., 40 (Suppl) (2007), pp. S83–S88
o
45.
o Doknic et al., 2002
o M. Doknic, S. Pekic, M. Zarkovic, M. Medic-Stojanoska, C. Dieguez, F. Casanueva, V. Popovic
o Dopaminergic tone and obesity: an insight from prolactinomas treated with bromocriptine
o Eur. J. Endocrinol., 147 (2002), pp. 77–84
o
46.
o Everitt and Robbins, 2005
o B.J. Everitt, T.W. Robbins
o Neural systems of reinforcement for drug addiction: From actions to habits to compulsion
o Nat. Neurosci., 8 (2005), pp. 1481–1489
o
47.
o Everitt et al., 2008
o B.J. Everitt, D. Belin, D. Economidou, Y. Pelloux, J.W. Dalley, T.W. Robbins
o Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction
o Philos. Trans. R. Soc. Lond. B Biol. Sci., 363 (2008), pp. 3125–3135
o
48.
o Farooqi et al., 2007
o I.S. Farooqi, E. Bullmore, J. Keogh, J. Gillard, S. O’Rahilly, P.C. Fletcher
o Leptin regulates striatal regions and human eating behavior
o Science, 317 (2007), p. 1355
o
49.
o Felsted et al., 2010
o J.A. Felsted, X. Ren, F. Chouinard-Decorte, D.M. Small
o Genetically determined differences in brain response to a primary food reward
o J. Neurosci., 30 (2010), pp. 2428–2432
o
50.
o Figlewicz et al., 2001
o D.P. Figlewicz, M.S. Higgins, S.B. Ng-Evans, P.J. Havel
o Leptin reverses sucrose-conditioned place preference in food-restricted rats
o Physiol. Behav., 73 (2001), pp. 229–234
o
51.
o Figlewicz et al., 2003
o D.P. Figlewicz, S.B. Evans, J. Murphy, M. Hoen, D.G. Baskin
o Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat
o Brain Res., 964 (2003), pp. 107–115
o
52.
o Fineberg et al., 2010
o N.A. Fineberg, M.N. Potenza, S.R. Chamberlain, H.A. Berlin, L. Menzies, A. Bechara, B.J. Sahakian, T.W. Robbins, E.T. Bullmore, E. Hollander
o Probing compulsive and impulsive behaviors, from animal models to endophenotypes: A narrative review
o Neuropsychopharmacology, 35 (2010), pp. 591–604
o
53.
o Finkelstein et al., 2005
o E.A. Finkelstein, C.J. Ruhm, K.M. Kosa
o Economic causes and consequences of obesity
o Annu. Rev. Public Health, 26 (2005), pp. 239–257
o
54.
o Flegal et al., 2010
o K.M. Flegal, M.D. Carroll, C.L. Ogden, L.R. Curtin
o Prevalence and trends in obesity among US adults, 1999-2008
o JAMA, 303 (2010), pp. 235–241
o
55.
o Foo and Mason, 2005
o H. Foo, P. Mason
o Sensory suppression during feeding
o Proc. Natl. Acad. Sci. USA, 102 (2005), pp. 16865–16869
o
56.
o Franken and Muris, 2005
o I.H. Franken, P. Muris
o Individual differences in reward sensitivity are related to food craving and relative body weight in healthy women
o Appetite, 45 (2005), pp. 198–201
o
57.
o Friedman et al., 2011
o A. Friedman, E. Lax, Y. Dikshtein, L. Abraham, Y. Flaumenhaft, E. Sudai, M. Ben-Tzion, G. Yadid
o Electrical stimulation of the lateral habenula produces an inhibitory effect on sucrose self-administration
o Neuropharmacology, 60 (2011), pp. 381–387
o
58.
o Fulton et al., 2000
o S. Fulton, B. Woodside, P. Shizgal
o Modulation of brain reward circuitry by leptin
o Science, 287 (2000), pp. 125–128
o
59.
o Fulton et al., 2006
o S. Fulton, P. Pissios, R.P. Manchon, L. Stiles, L. Frank, E.N. Pothos, E. Maratos-Flier, J.S. Flier
o Leptin regulation of the mesoaccumbens dopamine pathway
o Neuron, 51 (2006), pp. 811–822
o
60.
o Gao and Horvath, 2007
o Q. Gao, T.L. Horvath
o Neurobiology of feeding and energy expenditure
o Annu. Rev. Neurosci., 30 (2007), pp. 367–398
o
61.
o Gautier et al., 2000
o J.F. Gautier, K. Chen, A.D. Salbe, D. Bandy, R.E. Pratley, M. Heiman, E. Ravussin, E.M. Reiman, P.A. Tataranni
o Differential brain responses to satiation in obese and lean men
o Diabetes, 49 (2000), pp. 838–846
o
62.
o Geiger et al., 2008
o B.M. Geiger, G.G. Behr, L.E. Frank, A.D. Caldera-Siu, M.C. Beinfeld, E.G. Kokkotou, E.N. Pothos
o Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats
o FASEB J., 22 (2008), pp. 2740–2746
o
63.
o Geiger et al., 2009
o B.M. Geiger, M. Haburcak, N.M. Avena, M.C. Moyer, B.G. Hoebel, E.N. Pothos
o Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity
o Neuroscience, 159 (2009), pp. 1193–1199
o
64.
o Ghitza et al., 2006
o U.E. Ghitza, S.M. Gray, D.H. Epstein, K.C. Rice, Y. Shaham
o Neuropsychopharmacology
o The anxiogenic drug yohimbine reinstates palatable food seeking in a rat relapse model: A role of CRF(1) receptors, 33 (2006), pp. 2188–2196
o
65.
o Gluck et al., 2004
o M.E. Gluck, A. Geliebter, J. Hung, E. Yahav
o Cortisol, hunger, and desire to binge eat following a cold stress test in obese women with binge eating disorder
o Psychosom. Med., 66 (2004), pp. 876–881
o
66.
o Goldstone et al., 2009
o A.P. Goldstone, C.G. Prechtl de Hernandez, J.D. Beaver, K. Muhammed, C. Croese, G. Bell, G. Durighel, E. Hughes, A.D. Waldman, G. Frost, J.D. Bell
o Fasting biases brain reward systems towards high-calorie foods
o Eur. J. Neurosci., 30 (2009), pp. 1625–1635
o
67.
o Halaas et al., 1995
o J.L. Halaas, K.S. Gajiwala, M. Maffei, S.L. Cohen, B.T. Chait, D. Rabinowitz, R.L. Lallone, S.K. Burley, J.M. Friedman
o Weight-reducing effects of the plasma protein encoded by the obese gene
o Science, 269 (1995), pp. 543–546
o
68.
o Hamilton et al., 1995
o B.S. Hamilton, D. Paglia, A.Y. Kwan, M. Deitel
o Increased obese mRNA expression in omental fat cells from massively obese humans
o Nat. Med., 1 (1995), pp. 953–956
o
69.
o Hernandez and Hoebel, 1988
o L. Hernandez, B.G. Hoebel
o Food reward and cocaine increase extracellular dopamine in the nucleus accumbens as measured by microdialysis
o Life Sci., 42 (1988), pp. 1705–1712
o
70.
o Hill et al., 2003
o J.O. Hill, H.R. Wyatt, G.W. Reed, J.C. Peters
o Obesity and the environment: Where do we go from here?
o Science, 299 (2003), pp. 853–855
o
71.
o Hoebel, 1969
o B.G. Hoebel
o Feeding and self-stimulation
o Ann. N. Y. Acad. Sci., 157 (1969), pp. 758–778
o
72.
o Hoebel and Balagura, 1967
o B.G. Hoebel, S. Balagura
o Self-stimulation of the lateral hypothalamus modified by insulin and glucagon
o Physiol. Behav., 2 (1967), pp. 337–340
o
73.
o Hoebel and Teitelbaum, 1962
o B.G. Hoebel, P. Teitelbaum
o Hypothalamic control of feeding and self-stimulation
o Science, 135 (1962), pp. 375–377
o
74.
o Hoebel and Thompson, 1969
o B.G. Hoebel, R.D. Thompson
o Aversion to lateral hypothalamic stimulation caused by intragastric feeding or obesity
o J. Comp. Physiol. Psychol., 68 (1969), pp. 536–543
o
75.
o Hofmann et al., 2010
o W. Hofmann, G.M. van Koningsbruggen, W. Stroebe, S. Ramanathan, H. Aarts
o As pleasure unfolds: Hedonic responses to tempting food
o Psychol. Sci., 21 (2010), pp. 1863–1870
o
76.
o Holland and Gallagher, 2004
o P.C. Holland, M. Gallagher
o Amygdala-frontal interactions and reward expectancy
o Curr. Opin. Neurobiol., 14 (2004), pp. 148–155
o
77.
o Hollander et al., 2010
o J.A. Hollander, H.I. Im, A.L. Amelio, J. Kocerha, P. Bali, Q. Lu, D. Willoughby, C. Wahlestedt, M.D. Conkright, P.J. Kenny
o Striatal microRNA controls cocaine intake through CREB signalling
o Nature, 466 (2010), pp. 197–202
o
78.
o Hommel et al., 2006
o J.D. Hommel, R. Trinko, R.M. Sears, D. Georgescu, Z.W. Liu, X.B. Gao, J.J. Thurmon, M. Marinelli, R.J. DiLeone
o Leptin receptor signaling in midbrain dopamine neurons regulates feeding
o Neuron, 51 (2006), pp. 801–810
o
79.
o Imaizumi et al., 2001
o M. Imaizumi, M. Takeda, A. Suzuki, S. Sawano, T. Fushiki
o Preference for high-fat food in mice: Fried potatoes compared with boiled potatoes
o Appetite, 36 (2001), pp. 237–238
o
80.
o Jerlhag et al., 2006
o E. Jerlhag, E. Egecioglu, S.L. Dickson, M. Andersson, L. Svensson, J.A. Engel
o Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: Implications for its involvement in brain reward
o Addict. Biol., 11 (2006), pp. 45–54
o
81.
o Jerlhag et al., 2007
o E. Jerlhag, E. Egecioglu, S.L. Dickson, A. Douhan, L. Svensson, J.A. Engel
o Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens
o Addict. Biol., 12 (2007), pp. 6–16
o
82.
o Jhou et al., 2009
o T.C. Jhou, H.L. Fields, M.G. Baxter, C.B. Saper, P.C. Holland
o The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses
o Neuron, 61 (2009), pp. 786–800
o
83.
o Johnson and Kenny, 2010
o P.M. Johnson, P.J. Kenny
o Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats
o Nat. Neurosci., 13 (2010), pp. 635–641
o
84.
o Johnson et al., 1996
o P.I. Johnson, M.A. Parente, J.R. Stellar
o NMDA-induced lesions of the nucleus accumbens or the ventral pallidum increase the rewarding efficacy of food to deprived rats
o Brain Res., 722 (1996), pp. 109–117
o
85.
o Jönsson et al., 1999
o E.G. Jönsson, M.M. Nöthen, F. Grünhage, L. Farde, Y. Nakashima, P. Propping, G.C. Sedvall
o Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers
o Mol. Psychiatry, 4 (1999), pp. 290–296
o
86.
o Kalarchian et al., 2002
o M.A. Kalarchian, M.D. Marcus, G.T. Wilson, E.W. Labouvie, R.E. Brolin, L.B. LaMarca
o Binge eating among gastric bypass patients at long-term follow-up
o Obes. Surg., 12 (2002), pp. 270–275
o
87.
o Kandiah et al., 2006
o J. Kandiah, M. Yake, J. Jones, M. Meyer
o Stress influences appetite and comfort food preferences in college women
o Nutr. Res., 26 (2006), pp. 118–123
o
88.
o Karhunen et al., 1997
o L.J. Karhunen, R.I. Lappalainen, E.J. Vanninen, J.T. Kuikka, M.I. Uusitupa
o Regional cerebral blood flow during food exposure in obese and normal-weight women
o Brain, 120 (1997), pp. 1675–1684
o
89.
o Kasanetz et al., 2010
o F. Kasanetz, V. Deroche-Gamonet, N. Berson, E. Balado, M. Lafourcade, O. Manzoni, P.V. Piazza
o Transition to addiction is associated with a persistent impairment in synaptic plasticity
o Science, 328 (2010), pp. 1709–1712
o
90.
o Kelley et al., 1996
o A.E. Kelley, E.P. Bless, C.J. Swanson
o Investigation of the effects of opiate antagonists infused into the nucleus accumbens on feeding and sucrose drinking in rats
o J. Pharmacol. Exp. Ther., 278 (1996), pp. 1499–1507
o
91.
o Kelley et al., 2005
o A.E. Kelley, B.A. Baldo, W.E. Pratt, M.J. Will
o Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward
o Physiol. Behav., 86 (2005), pp. 773–795
o
92.
o Kenny et al., 2006
o P.J. Kenny, S.A. Chen, O. Kitamura, A. Markou, G.F. Koob
o Conditioned withdrawal drives heroin consumption and decreases reward sensitivity
o J. Neurosci., 26 (2006), pp. 5894–5900
o
93.
o Kim et al., 2010
o K.S. Kim, Y.R. Yoon, H.J. Lee, S. Yoon, S.Y. Kim, S.W. Shin, J.J. An, M.S. Kim, S.Y. Choi, W. Sun, J.H. Baik
o Enhanced hypothalamic leptin signaling in mice lacking dopamine D2 receptors
o J. Biol. Chem., 285 (2010), pp. 8905–8917
o
94.
o Klein et al., 2007
o T.A. Klein, J. Neumann, M. Reuter, J. Hennig, D.Y. von Cramon, M. Ullsperger
o Genetically determined differences in learning from errors
o Science, 318 (2007), pp. 1642–1645
o
95.
o Kojima et al., 1999
o M. Kojima, H. Hosoda, Y. Date, M. Nakazato, H. Matsuo, K. Kangawa
o Ghrelin is a growth-hormone-releasing acylated peptide from stomach
o Nature, 402 (1999), pp. 656–660
o
96.
o Kok et al., 2006
o P. Kok, F. Roelfsema, M. Frölich, J. van Pelt, A.E. Meinders, H. Pijl
o Activation of dopamine D2 receptors lowers circadian leptin concentrations in obese women
o J. Clin. Endocrinol. Metab., 91 (2006), pp. 3236–3240
o
97.
o Koob, 2010
o G.F. Koob
o The role of CRF and CRF-related peptides in the dark side of addiction
o Brain Res., 1314 (2010), pp. 3–14
o
98.
o Koob and Le Moal, 2008
o G.F. Koob, M. Le Moal
o Addiction and the brain antireward system
o Annu. Rev. Psychol., 59 (2008), pp. 29–53
o
99.
o Koob and Volkow, 2010
o G.F. Koob, N.D. Volkow
o Neurocircuitry of addiction
o Neuropsychopharmacology, 35 (2010), pp. 217–238
o
100.
o Koob and Zorrilla, 2010
o G.F. Koob, E.P. Zorrilla
o Neurobiological mechanisms of addiction: Focus on corticotropin-releasing factor
o Curr. Opin. Investig. Drugs, 11 (2010), pp. 63–71
o
101.
o Kringelbach et al., 2003
o M.L. Kringelbach, J. O’Doherty, E.T. Rolls, C. Andrews
o Activation of the human orbitofrontal cortex to a liquid food stimulus is correlated with its subjective pleasantness
o Cereb. Cortex, 13 (2003), pp. 1064–1071
o
102.
o Krügel et al., 2003
o U. Krügel, T. Schraft, H. Kittner, W. Kiess, P. Illes
o Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin
o Eur. J. Pharmacol., 482 (2003), pp. 185–187
o
103.
o la Fleur, 2006
o S.E. la Fleur
o The effects of glucocorticoids on feeding behavior in rats
o Physiol. Behav., 89 (2006), pp. 110–114
o
104.
o LaBar et al., 2001
o K.S. LaBar, D.R. Gitelman, T.B. Parrish, Y.H. Kim, A.C. Nobre, M.M. Mesulam
o Hunger selectively modulates corticolimbic activation to food stimuli in humans
o Behav. Neurosci., 115 (2001), pp. 493–500
o
105.
o Latagliata et al., 2010
o E.C. Latagliata, E. Patrono, S. Puglisi-Allegra, R. Ventura
o Food seeking in spite of harmful consequences is under prefrontal cortical noradrenergic control
o BMC Neurosci., 11 (2010), p. 15
o
106.
o Lawford et al., 2000
o B.R. Lawford, R.M. Young, E.P. Noble, J. Sargent, J. Rowell, S. Shadforth, X. Zhang, T. Ritchie
o The D(2) dopamine receptor A(1) allele and opioid dependence: association with heroin use and response to methadone treatment
o Am. J. Med. Genet., 96 (2000), pp. 592–598
o
107.
o Leinninger et al., 2009
o G.M. Leinninger, Y.H. Jo, R.L. Leshan, G.W. Louis, H. Yang, J.G. Barrera, H. Wilson, D.M. Opland, M.A. Faouzi, Y. Gong et al.
o Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding
o Cell Metab., 10 (2009), pp. 89–98
o
108.
o Lenoir et al., 2007
o M. Lenoir, F. Serre, L. Cantin, S.H. Ahmed
o Intense sweetness surpasses cocaine reward
o PLoS ONE, 2 (2007), p. e698 http://dx.doi.org/10.1371/journal.pone.0000698
o
109.
o Leshan et al., 2010
o R.L. Leshan, D.M. Opland, G.W. Louis, G.M. Leinninger, C.M. Patterson, C.J. Rhodes, H. Münzberg, M.G. Myers Jr.
o Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala
o J. Neurosci., 30 (2010), pp. 5713–5723
o
110.
o Louis et al., 2010
o G.W. Louis, G.M. Leinninger, C.J. Rhodes, M.G. Myers Jr.
o Direct innervation and modulation of orexin neurons by lateral hypothalamic LepRb neurons
o J. Neurosci., 30 (2010), pp. 11278–11287
o
111.
o Luppino et al., 2010
o F.S. Luppino, L.M. de Wit, P.F. Bouvy, T. Stijnen, P. Cuijpers, B.W. Penninx, F.G. Zitman
o Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies
o Arch. Gen. Psychiatry, 67 (2010), pp. 220–229
o
112.
o Lutter and Nestler, 2009
o M. Lutter, E.J. Nestler
o Homeostatic and hedonic signals interact in the regulation of food intake
o J. Nutr., 139 (2009), pp. 629–632
o
113.
o Macht and Mueller, 2007
o M. Macht, J. Mueller
o Immediate effects of chocolate on experimentally induced mood states
o Appetite, 49 (2007), pp. 667–674
o
114.
o Maldonado-Irizarry and Kelley, 1995
o C.S. Maldonado-Irizarry, A.E. Kelley
o Excitotoxic lesions of the core and shell subregions of the nucleus accumbens differentially disrupt body weight regulation and motor activity in rat
o Brain Res. Bull., 38 (1995), pp. 551–559
o
115.
o Maldonado-Irizarry et al., 1995
o C.S. Maldonado-Irizarry, C.J. Swanson, A.E. Kelley
o Glutamate receptors in the nucleus accumbens shell control feeding behavior via the lateral hypothalamus
o J. Neurosci., 15 (1995), pp. 6779–6788
o
116.
o Malik et al., 2008
o S. Malik, F. McGlone, D. Bedrossian, A. Dagher
o Ghrelin modulates brain activity in areas that control appetitive behavior
o Cell Metab., 7 (2008), pp. 400–409
o
117.
o Man et al., 2009
o M.S. Man, H.F. Clarke, A.C. Roberts
o The role of the orbitofrontal cortex and medial striatum in the regulation of prepotent responses to food rewards
o Cereb. Cortex, 19 (2009), pp. 899–906
o
118.
o Margules and Olds, 1962
o D.L. Margules, J. Olds
o Identical “feeding” and “rewarding” systems in the lateral hypothalamus of rats
o Science, 135 (1962), pp. 374–375
o
119.
o Markou and Frank, 1987
o A. Markou, R.A. Frank
o The effect of operant and electrode placement on self-stimulation train duration response functions
o Physiol. Behav., 41 (1987), pp. 303–308
o
120.
o Markou and Koob, 1991
o A. Markou, G.F. Koob
o Postcocaine anhedonia. An animal model of cocaine withdrawal
o Neuropsychopharmacology, 4 (1991), pp. 17–26
o
121.
o Mastronardi et al., 2001
o C.A. Mastronardi, W.H. Yu, V.K. Srivastava, W.L. Dees, S.M. McCann
o Lipopolysaccharide-induced leptin release is neurally controlled
o Proc. Natl. Acad. Sci. USA, 98 (2001), pp. 14720–14725
o
122.
o Matheny et al., 2011
o M. Matheny, A. Shapiro, N. Tumer, P.J. Scarpace
o Region-specific diet-induced and leptin-induced cellular leptin resistance includes the ventral tegmental area in rats
o Neuropharmacology (2011) http://dx.doi.org/10.1016/j.neuropharm.2010.11.002 in press. Published online November 5, 2010
o
123.
o Matsumoto and Hikosaka, 2007
o M. Matsumoto, O. Hikosaka
o Lateral habenula as a source of negative reward signals in dopamine neurons
o Nature, 447 (2007), pp. 1111–1115
o
124.
o Morton et al., 2006
o G.J. Morton, D.E. Cummings, D.G. Baskin, G.S. Barsh, M.W. Schwartz
o Central nervous system control of food intake and body weight
o Nature, 443 (2006), pp. 289–295
o
125.
o Mount and Hoebel, 1967
o G. Mount, B.G. Hoebel
o Lateral hypothalamic self-stimulation: Self-determined threshold increased by food intake
o Psychon. Sci., 9 (1967), pp. 265–266
o
126.
o Nair et al., 2011
o S.G. Nair, B.M. Navarre, C. Cifani, C.L. Pickens, J.M. Bossert, Y. Shaham
o Role of dorsal medial prefrontal cortex dopamine D1-family receptors in relapse to high-fat food seeking induced by the anxiogenic drug yohimbine
o Neuropsychopharmacology, 36 (2011), pp. 497–510
o
127.
o Nakazato et al., 2001
o M. Nakazato, N. Murakami, Y. Date, M. Kojima, H. Matsuo, K. Kangawa, S. Matsukura
o A role for ghrelin in the central regulation of feeding
o Nature, 409 (2001), pp. 194–198
o
128.
o Naqvi and Bechara, 2009
o N.H. Naqvi, A. Bechara
o The hidden island of addiction: The insula
o Trends Neurosci., 32 (2009), pp. 56–67
o
129.
o Neville et al., 2004
o M.J. Neville, E.C. Johnstone, R.T. Walton
o Identification and characterization of ANKK1: A novel kinase gene closely linked to DRD2 on chromosome band 11q23.1
o Hum. Mutat., 23 (2004), pp. 540–545
o
130.
o Nirenberg and Waters, 2006
o M.J. Nirenberg, C. Waters
o Compulsive eating and weight gain related to dopamine agonist use
o Mov. Disord., 21 (2006), pp. 524–529
o
131.
o Noble, 2000
o E.P. Noble
o Addiction and its reward process through polymorphisms of the D2 dopamine receptor gene: A review
o Eur. Psychiatry, 15 (2000), pp. 79–89
o
132.
o Noble et al., 1993
o E.P. Noble, K. Blum, M.E. Khalsa, T. Ritchie, A. Montgomery, R.C. Wood, R.J. Fitch, T. Ozkaragoz, P.J. Sheridan, M.D. Anglin et al.
o Allelic association of the D2 dopamine receptor gene with cocaine dependence
o Drug Alcohol Depend., 33 (1993), pp. 271–285
o
133.
o Noble et al., 2000
o E.P. Noble, X. Zhang, T.L. Ritchie, R.S. Sparkes
o Haplotypes at the DRD2 locus and severe alcoholism
o Am. J. Med. Genet., 96 (2000), pp. 622–631
o
134.
o O’Connor et al., 2008
o D.B. O’Connor, F. Jones, M. Conner, B. McMillan, E. Ferguson
o Effects of daily hassles and eating style on eating behavior
o Health Psychol., 27 (1, Suppl) (2008), pp. S20–S31
o
135.
o O’Doherty et al., 2002
o J.P. O’Doherty, R. Deichmann, H.D. Critchley, R.J. Dolan
o Neural responses during anticipation of a primary taste reward
o Neuron, 33 (2002), pp. 815–826
o
136.
o O’Rahilly, 2009
o S. O’Rahilly
o Human genetics illuminates the paths to metabolic disease
o Nature, 462 (2009), pp. 307–314
o
137.
o Oswald et al., 2010
o K.D. Oswald, D.L. Murdaugh, V.L. King, M.M. Boggiano
o Motivation for palatable food despite consequences in an animal model of binge eating
o Int. J. Eat. Disord. (2010) http://dx.doi.org/10.1002/eat.20808 in press. Published online February 22, 2010
o
138.
o Park and Carr, 1998
o T.H. Park, K.D. Carr
o Neuroanatomical patterns of fos-like immunoreactivity induced by a palatable meal and meal-paired environment in saline- and naltrexone-treated rats
o Brain Res., 805 (1998), pp. 169–180
o
139.
o Peciña and Berridge, 2005
o S. Peciña, K.C. Berridge
o Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness?
o J. Neurosci., 25 (2005), pp. 11777–11786
o
140.
o Peciña et al., 2006a
o S. Peciña, J. Schulkin, K.C. Berridge
o Nucleus accumbens corticotropin-releasing factor increases cue-triggered motivation for sucrose reward: Paradoxical positive incentive effects in stress?
o BMC Biol., 4 (2006), p. 8
o
141.
o Peciña et al., 2006b
o S. Peciña, K.S. Smith, K.C. Berridge
o Hedonic hot spots in the brain
o Neuroscientist, 12 (2006), pp. 500–511
o
142.
o Pecoraro et al., 2004
o N. Pecoraro, F. Reyes, F. Gomez, A. Bhargava, M.F. Dallman
o Chronic stress promotes palatable feeding, which reduces signs of stress: Feedforward and feedback effects of chronic stress
o Endocrinology, 145 (2004), pp. 3754–3762
o
143.
o Pelchat et al., 2004
o M.L. Pelchat, A. Johnson, R. Chan, J. Valdez, J.D. Ragland
o Images of desire: Food-craving activation during fMRI
o Neuroimage, 23 (2004), pp. 1486–1493
o
144.
o Pelleymounter et al., 1995
o M.A. Pelleymounter, M.J. Cullen, M.B. Baker, R. Hecht, D. Winters, T. Boone, F. Collins
o Effects of the obese gene product on body weight regulation in ob/ob mice
o Science, 269 (1995), pp. 540–543
o
145.
o Pelloux et al., 2007
o Y. Pelloux, B.J. Everitt, A. Dickinson
o Compulsive drug seeking by rats under punishment: Effects of drug taking history
o Psychopharmacology (Berl.), 194 (2007), pp. 127–137
o
146.
o Perello et al., 2010
o M. Perello, I. Sakata, S. Birnbaum, J.C. Chuang, S. Osborne-Lawrence, S.A. Rovinsky, J. Woloszyn, M. Yanagisawa, M. Lutter, J.M. Zigman
o Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner
o Biol. Psychiatry, 67 (2010), pp. 880–886
o
147.
o Petrovich et al., 2009
o G.D. Petrovich, C.A. Ross, P. Mody, P.C. Holland, M. Gallagher
o Central, but not basolateral, amygdala is critical for control of feeding by aversive learned cues
o J. Neurosci., 29 (2009), pp. 15205–15212
o
148.
o Pfaffly et al., 2010
o J. Pfaffly, M. Michaelides, G.J. Wang, J.E. Pessin, N.D. Volkow, P.K. Thanos
o Leptin increases striatal dopamine D2 receptor binding in leptin-deficient obese (ob/ob) mice
o Synapse, 64 (2010), pp. 503–510
o
149.
o Puhl et al., 2008
o R.M. Puhl, C.A. Moss-Racusin, M.B. Schwartz, K.D. Brownell
o Weight stigmatization and bias reduction: Perspectives of overweight and obese adults
o Health Educ. Res., 23 (2008), pp. 347–358
o
150.
o Rada et al., 2010
o P. Rada, M.E. Bocarsly, J.R. Barson, B.G. Hoebel, S.F. Leibowitz
o Reduced accumbens dopamine in Sprague-Dawley rats prone to overeating a fat-rich diet
o Physiol. Behav., 101 (2010), pp. 394–400
o
151.
o Ritchie and Noble, 2003
o T. Ritchie, E.P. Noble
o Association of seven polymorphisms of the D2 dopamine receptor gene with brain receptor-binding characteristics
o Neurochem. Res., 28 (2003), pp. 73–82
o
152.
o Roberto et al., 2010
o M. Roberto, M.T. Cruz, N.W. Gilpin, V. Sabino, P. Schweitzer, M. Bajo, P. Cottone, S.G. Madamba, D.G. Stouffer, E.P. Zorrilla et al.
o Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence
o Biol. Psychiatry, 67 (2010), pp. 831–839
o
153.
o Roitman et al., 2004
o M.F. Roitman, G.D. Stuber, P.E. Phillips, R.M. Wightman, R.M. Carelli
o Dopamine operates as a subsecond modulator of food seeking
o J. Neurosci., 24 (2004), pp. 1265–1271
o
154.
o Roitman et al., 2008
o M.F. Roitman, R.A. Wheeler, R.M. Wightman, R.M. Carelli
o Real-time chemical responses in the nucleus accumbens differentiate rewarding and aversive stimuli
o Nat. Neurosci., 11 (2008), pp. 1376–1377
o
155.
o Rolls, 2008
o E.T. Rolls
o Functions of the orbitofrontal and pregenual cingulate cortex in taste, olfaction, appetite and emotion
o Acta Physiol. Hung., 95 (2008), pp. 131–164
o
156.
o Rolls, 2010
o E.T. Rolls
o Taste, olfactory and food texture reward processing in the brain and obesity
o Int. J. Obes. (Lond.) (2010) http://dx.doi.org/10.1038/ijo.2010.155 in press. Published online August 3, 2010
o
157.
o Rolls et al., 1983
o E.T. Rolls, B.J. Rolls, E.A. Rowe
o Sensory-specific and motivation-specific satiety for the sight and taste of food and water in man
o Physiol. Behav., 30 (1983), pp. 185–192
o
158.
o Roseberry et al., 2007
o A.G. Roseberry, T. Painter, G.P. Mark, J.T. Williams
o Decreased vesicular somatodendritic dopamine stores in leptin-deficient mice
o J. Neurosci., 27 (2007), pp. 7021–7027
o
159.
o Rothemund et al., 2007
o Y. Rothemund, C. Preuschhof, G. Bohner, H.C. Bauknecht, R. Klingebiel, H. Flor, B.F. Klapp
o Differential activation of the dorsal striatum by high-calorie visual food stimuli in obese individuals
o Neuroimage, 37 (2007), pp. 410–421
o
160.
o Salas et al., 2010
o R. Salas, P. Baldwin, M. de Biasi, P.R. Montague
o BOLD responses to negative reward prediction errors in human habenula
o Front. Hum. Neurosci., 4 (2010), p. 36
o
161.
o Saper et al., 2002
o C.B. Saper, T.C. Chou, J.K. Elmquist
o The need to feed: homeostatic and hedonic control of eating
o Neuron, 36 (2002), pp. 199–211
o
162.
o Saunders, 2001
o R. Saunders
o Compulsive eating and gastric bypass surgery: What does hunger have to do with it?
o Obes. Surg., 11 (2001), pp. 757–761
o
163.
o Scarpace et al., 2010
o P.J. Scarpace, M. Matheny, Y. Zhang
o Wheel running eliminates high-fat preference and enhances leptin signaling in the ventral tegmental area
o Physiol. Behav., 100 (2010), pp. 173–179
o
164.
o Schiltz et al., 2007
o C.A. Schiltz, Q.Z. Bremer, C.F. Landry, A.E. Kelley
o Food-associated cues alter forebrain functional connectivity as assessed with immediate early gene and proenkephalin expression
o BMC Biol., 5 (2007), p. 16
o
165.
o Schur et al., 2009
o E.A. Schur, N.M. Kleinhans, J. Goldberg, D. Buchwald, M.W. Schwartz, K. Maravilla
o Activation in brain energy regulation and reward centers by food cues varies with choice of visual stimulus
o Int. J. Obes. (Lond.), 33 (2009), pp. 653–661
o
166.
o Sclafani et al., 1998
o A. Sclafani, R.J. Bodnar, A.R. Delamater
o Pharmacology of food conditioned preferences
o Appetite, 31 (1998), p. 406
o
167.
o Sescousse et al., 2010
o G. Sescousse, J. Redouté, J.C. Dreher
o The architecture of reward value coding in the human orbitofrontal cortex
o J. Neurosci., 30 (2010), pp. 13095–13104
o
168.
o Shomaker et al., 2010
o L.B. Shomaker, M. Tanofsky-Kraff, J.M. Zocca, A. Courville, M. Kozlosky, K.M. Columbo, L.E. Wolkoff, S.M. Brady, M.K. Crocker, A.H. Ali et al.
o Eating in the absence of hunger in adolescents: Intake after a large-array meal compared with that after a standardized meal
o Am. J. Clin. Nutr., 92 (2010), pp. 697–703
o
169.
o Simmons et al., 2005
o W.K. Simmons, A. Martin, L.W. Barsalou
o Pictures of appetizing foods activate gustatory cortices for taste and reward
o Cereb. Cortex, 15 (2005), pp. 1602–1608
o
170.
o Small, 2010
o D.M. Small
o Taste representation in the human insula
o Brain Struct. Funct., 214 (2010), pp. 551–561
o
171.
o Small et al., 2003
o D.M. Small, M. Jones-Gotman, A. Dagher
o Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers
o Neuroimage, 19 (2003), pp. 1709–1715
o
172.
o Smith and Berridge, 2007
o K.S. Smith, K.C. Berridge
o Opioid limbic circuit for reward: interaction between hedonic hotspots of nucleus accumbens and ventral pallidum
o J. Neurosci., 27 (2007), pp. 1594–1605
o
173.
o Söderpalm and Berridge, 2000
o A.H. Söderpalm, K.C. Berridge
o Food intake after diazepam, morphine or muscimol: Microinjections In the nucleus accumbens shell
o Pharmacol. Biochem. Behav., 66 (2000), pp. 429–434
o
174.
o Stice et al., 2008a
o E. Stice, S. Spoor, C. Bohon, D.M. Small
o Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele
o Science, 322 (2008), pp. 449–452
o
175.
o Stice et al., 2008b
o E. Stice, S. Spoor, C. Bohon, M.G. Veldhuizen, D.M. Small
o Relation of reward from food intake and anticipated food intake to obesity: A functional magnetic resonance imaging study
o J. Abnorm. Psychol., 117 (2008), pp. 924–935
o
176.
o Stice et al., 2010a
o E. Stice, S. Yokum, K. Blum, C. Bohon
o Weight gain is associated with reduced striatal response to palatable food
o J. Neurosci., 30 (2010), pp. 13105–13109
o
177.
o Stice et al., 2010b
o E. Stice, S. Yokum, C. Bohon, N. Marti, A. Smolen
o Reward circuitry responsivity to food predicts future increases in body mass: Moderating effects of DRD2 and DRD4
o Neuroimage, 50 (2010), pp. 1618–1625
o
178.
o Stoeckel, 2010
o Stoeckel, L.E. (2010). The Goldilocks Principle of Obesity. Scientific American. June 8, 2010. http://www.scientificamerican.com/article.cfm?id=the-goldilocks-principle-obesity.
o
179.
o Stratford and Kelley, 1997
o T.R. Stratford, A.E. Kelley
o GABA in the nucleus accumbens shell participates in the central regulation of feeding behavior
o J. Neurosci., 17 (1997), pp. 4434–4440
o
180.
o Stratford and Kelley, 1999
o T.R. Stratford, A.E. Kelley
o Evidence of a functional relationship between the nucleus accumbens shell and lateral hypothalamus subserving the control of feeding behavior
o J. Neurosci., 19 (1999), pp. 11040–11048
o
181.
o Suarez and Gallup, 1981
o S.D. Suarez, G.G.J. Gallup
o An ethological analysis of open-field behavior in rats and mice
o Learn. Motiv., 12 (1981), pp. 342–363
o
182.
o Sunday et al., 1983
o S.R. Sunday, S.A. Sanders, G. Collier
o Palatability and meal patterns
o Physiol. Behav., 30 (1983), pp. 915–918
o
183.
o Swinburn et al., 2009
o B. Swinburn, G. Sacks, E. Ravussin
o Increased food energy supply is more than sufficient to explain the US epidemic of obesity
o Am. J. Clin. Nutr., 90 (2009), pp. 1453–1456
o
184.
o Taha and Fields, 2005
o S.A. Taha, H.L. Fields
o Encoding of palatability and appetitive behaviors by distinct neuronal populations in the nucleus accumbens
o J. Neurosci., 25 (2005), pp. 1193–1202
o
185.
o Taha et al., 2009
o S.A. Taha, Y. Katsuura, D. Noorvash, A. Seroussi, H.L. Fields
o Convergent, not serial, striatal and pallidal circuits regulate opioid-induced food intake
o Neuroscience, 161 (2009), pp. 718–733
o
186.
o Teegarden and Bale, 2007
o S.L. Teegarden, T.L. Bale
o Decreases in dietary preference produce increased emotionality and risk for dietary relapse
o Biol. Psychiatry, 61 (2007), pp. 1021–1029
o
187.
o Teegarden et al., 2009
o S.L. Teegarden, A.N. Scott, T.L. Bale
o Early life exposure to a high fat diet promotes long-term changes in dietary preferences and central reward signaling
o Neuroscience, 162 (2009), pp. 924–932
o
188.
o Thanos et al., 2008
o P.K. Thanos, M. Michaelides, Y.K. Piyis, G.J. Wang, N.D. Volkow
o Food restriction markedly increases dopamine D2 receptor (D2R) in a rat model of obesity as assessed with in-vivo muPET imaging ([11C] raclopride) and in-vitro ([3H] spiperone) autoradiography
o Synapse, 62 (2008), pp. 50–61
o
189.
o Thompson and Swanson, 2010
o R.H. Thompson, L.W. Swanson
o Hypothesis-driven structural connectivity analysis supports network over hierarchical model of brain architecture
o Proc. Natl. Acad. Sci. USA, 107 (2010), pp. 15235–15239
o
190.
o Vanderschuren and Everitt, 2004
o L.J. Vanderschuren, B.J. Everitt
o Drug seeking becomes compulsive after prolonged cocaine self-administration
o Science, 305 (2004), pp. 1017–1019
o
191.
o Vendruscolo et al., 2010a
o L.F. Vendruscolo, A.B. Gueye, M. Darnaudéry, S.H. Ahmed, M. Cador
o Sugar overconsumption during adolescence selectively alters motivation and reward function in adult rats
o PLoS ONE, 5 (2010), p. e9296 http://dx.doi.org/10.1371/journal.pone.0009296
o
192.
o Vendruscolo et al., 2010b
o L.F. Vendruscolo, A.B. Gueye, J.C. Vendruscolo, K.J. Clemens, P. Mormède, M. Darnaudéry, M. Cador
o Reduced alcohol drinking in adult rats exposed to sucrose during adolescence
o Neuropharmacology, 59 (2010), pp. 388–394
o
193.
o Volkow and O’Brien, 2007
o N.D. Volkow, C.P. O’Brien
o Issues for DSM-V: Should obesity be included as a brain disorder?
o Am. J. Psychiatry, 164 (2007), pp. 708–710
o
194.
o Volkow and Wise, 2005
o N.D. Volkow, R.A. Wise
o How can drug addiction help us understand obesity?
o Nat. Neurosci., 8 (2005), pp. 555–560
o
195.
o Volkow et al., 2009
o N.D. Volkow, G.J. Wang, F. Telang, J.S. Fowler, R.Z. Goldstein, N. Alia-Klein, J. Logan, C. Wong, P.K. Thanos, Y. Ma, K. Pradhan
o Inverse association between BMI and prefrontal metabolic activity in healthy adults
o Obesity (Silver Spring), 17 (2009), pp. 60–65
o
196.
o Vucetic and Reyes, 2010
o Z. Vucetic, T.M. Reyes
o Central dopaminergic circuitry controlling food intake and reward: implications for the regulation of obesity
o Wiley Interdiscip. Rev. Syst. Biol. Med., 2 (2010), pp. 577–593
o
197.
o Wang et al., 2001
o G.J. Wang, N.D. Volkow, J. Logan, N.R. Pappas, C.T. Wong, W. Zhu, N. Netusil, J.S. Fowler
o Brain dopamine and obesity
o Lancet, 357 (2001), pp. 354–357
o
198.
o Wang et al., 2002
o G.J. Wang, N.D. Volkow, J.S. Fowler
o The role of dopamine in motivation for food in humans: Implications for obesity
o Expert Opin. Ther. Targets, 6 (2002), pp. 601–609
o
199.
o Wang et al., 2004a
o G.J. Wang, N.D. Volkow, F. Telang, M. Jayne, J. Ma, M. Rao, W. Zhu, C.T. Wong, N.R. Pappas, A. Geliebter, J.S. Fowler
o Exposure to appetitive food stimuli markedly activates the human brain
o Neuroimage, 21 (2004), pp. 1790–1797
o
200.
o Wang et al., 2004b
o G.J. Wang, N.D. Volkow, P.K. Thanos, J.S. Fowler
o Similarity between obesity and drug addiction as assessed by neurofunctional imaging: a concept review
o J. Addict. Dis., 23 (2004), pp. 39–53
o
201.
o Wang et al., 2008
o G.J. Wang, D. Tomasi, W. Backus, R. Wang, F. Telang, A. Geliebter, J. Korner, A. Bauman, J.S. Fowler, P.K. Thanos, N.D. Volkow
o Gastric distention activates satiety circuitry in the human brain
o Neuroimage, 39 (2008), pp. 1824–1831
o
202.
o Wassum et al., 2009
o K.M. Wassum, S.B. Ostlund, N.T. Maidment, B.W. Balleine
o Distinct opioid circuits determine the palatability and the desirability of rewarding events
o Proc. Natl. Acad. Sci. USA, 106 (2009), pp. 12512–12517
o
203.
o Wassum et al., 2011
o K.M. Wassum, I.C. Cely, B.W. Balleine, N.T. Maidment
o μ-Opioid receptor activation in the basolateral amygdala mediates the learning of increases but not decreases in the incentive value of a food reward
o J. Neurosci., 31 (2011), pp. 1591–1599
o
204.
o Wilkinson and Peele, 1962
o H.A. Wilkinson, T.L. Peele
o Modification of intracranial self-stimulation by hunger satiety
o Am. J. Physiol., 203 (1962), pp. 537–540
o
205.
o Will et al., 2003
o M.J. Will, E.B. Franzblau, A.E. Kelley
o Nucleus accumbens mu-opioids regulate intake of a high-fat diet via activation of a distributed brain network
o J. Neurosci., 23 (2003), pp. 2882–2888
o
206.
o Wilson et al., 2008
o M.E. Wilson, J. Fisher, A. Fischer, V. Lee, R.B. Harris, T.J. Bartness
o Quantifying food intake in socially housed monkeys: social status effects on caloric consumption
o Physiol. Behav., 94 (2008), pp. 586–594
o Article | PDF (553 K) |
View Record in Scopus
| Cited By in Scopus (22)
207.
o Yurcisin et al., 2009
o B.M. Yurcisin, M.M. Gaddor, E.J. DeMaria
o Obesity and bariatric surgery
o Clin. Chest Med., 30 (2009), pp. 539–553, ix
o
208.
o Zhang et al., 1998
o M. Zhang, B.A. Gosnell, A.E. Kelley
o Intake of high-fat food is selectively enhanced by mu opioid receptor stimulation within the nucleus accumbens
o J. Pharmacol. Exp. Ther., 285 (1998), pp. 908–914
o View Record in Scopus
| Cited By in Scopus (148)
209.
o Zheng et al., 2007
o H. Zheng, L.M. Patterson, H.R. Berthoud
o Orexin signaling in the ventral tegmental area is required for high-fat appetite induced by opioid stimulation of the nucleus accumbens
o J. Neurosci., 27 (2007), pp. 11075–11082
o View Record in Scopus
| Cited By in Scopus (64)
210.
o Zheng et al., 2009
o H. Zheng, N.R. Lenard, A.C. Shin, H.R. Berthoud
o Appetite control and energy balance regulation in the modern world: Reward-driven brain overrides repletion signals
o Int. J. Obes. (Lond.), 33 (Suppl 2) (2009), pp. S8–S13
o View Record in Scopus
| Cited By in Scopus (34)