Dialoghi Clin Neurosci. 2013 Dec;15(4):431-43.
Astratto
Nonostante l'importanza di numerosi fattori psicosociali, al suo interno, la tossicodipendenza coinvolge un processo biologico: la capacità di esposizione ripetuta a una droga di abuso per indurre cambiamenti in un cervello vulnerabile che guida la ricerca compulsiva e l'assunzione di droghe e la perdita di controllo sull'uso di droghe, che definisce uno stato di dipendenza. Qui, esaminiamo i tipi di adattamenti molecolari e cellulari che si verificano in specifiche regioni del cervello per mediare le anomalie comportamentali associate alla dipendenza. Questi includono alterazioni nell'espressione genica raggiunte in parte attraverso meccanismi epigenetici, plasticità nel funzionamento neurofisiologico di neuroni e sinapsi e plasticità associata nella morfologia neuronale e sinaptica mediata in parte da alterata segnalazione del fattore neurotrofico. Ognuno di questi tipi di modifiche indotte da farmaci può essere visto come una forma di "memoria cellulare o molecolare" Inoltre, è sorprendente che la maggior parte delle forme di plasticità legate alla dipendenza siano molto simili ai tipi di plasticità che sono stati associati a forme più classiche di "memoria comportamentale", forse riflettendo il repertorio finito di meccanismi adattivi disponibili per i neuroni di fronte con sfide ambientali. Infine, gli adattamenti molecolari e cellulari legati alla dipendenza coinvolgono la maggior parte delle stesse regioni cerebrali che mediano forme più classiche di memoria, coerentemente con l'idea che le memorie anormali sono driver importanti delle sindromi da dipendenza. L'obiettivo di questi studi che mirano a spiegare le basi molecolari e cellulari della tossicodipendenza è quello di sviluppare alla fine test diagnostici basati sulla biologia, nonché trattamenti più efficaci per i disturbi da dipendenza.
Introduzione
La tossicodipendenza, che può essere definita come ricerca e assunzione compulsiva di farmaci nonostante le orrende conseguenze o la perdita del controllo sull'uso di droghe, è causata da cambiamenti duraturi indotti da farmaci che si verificano in alcune regioni del cervello.1 Solo alcuni individui, tuttavia, soccombono alla dipendenza di fronte all'esposizione ripetuta ai farmaci, mentre altri sono in grado di usare casualmente una droga e di sfuggire a una sindrome da dipendenza. I fattori genetici rappresentano all'incirca il 50% di questa variabilità individuale nella vulnerabilità della tossicodipendenza e questo grado di ereditabilità è vero per tutte le principali classi di farmaci che provocano dipendenza, inclusi stimolanti, oppiacei, alcol, nicotina e cannabinoidi.2 Non è stato ancora possibile identificare la maggior parte dei geni che compongono questo rischio genetico, probabilmente a causa del coinvolgimento di forse centinaia di variazioni genetiche che si sommano in un singolo individuo per conferire vulnerabilità alla dipendenza (o, in altri individui, resistenza).
L'altro 50% del rischio di dipendenza è dovuto a una serie di fattori ambientali, che si verificano nel corso della vita, che interagiscono con la composizione genetica di un individuo per renderlo vulnerabile alla dipendenza in misura maggiore o minore. Diversi tipi di fattori ambientali sono stati implicati nella dipendenza, compresi gli stress psicosociali, ma di gran lunga il fattore più potente è l'esposizione stessa a una droga d'abuso. Alcuni farmaci "gateway", in particolare la nicotina, hanno dimostrato di aumentare la propria vulnerabilità a una dipendenza da un'altra droga.3 Inoltre, vi è una crescente evidenza che, nonostante una serie di rischi genetici per la dipendenza in tutta la popolazione, l'esposizione a dosi sufficientemente elevate di un farmaco per lunghi periodi di tempo può trasformare qualcuno che ha un carico genetico relativamente inferiore in un tossicodipendente..4
Negli ultimi due decenni sono stati fatti grandi progressi nell'identificazione sia delle regioni discrete del cervello che sono importanti nella mediazione di una sindrome da dipendenza, sia dei tipi di cambiamenti a livello molecolare e cellulare che i farmaci inducono in queste regioni a sottostare a aspetti chiave di dipendenza.1,5 Il circuito che ha ricevuto maggiore attenzione è indicato come il sistema mesolimbico della dopamina, che coinvolge i neuroni della dopamina nell'area tegmentale ventrale (VTA) del mesencefalo che innervano i neuroni spinosi medi nel nucleo accumbens (NAc, una parte dello striato ventrale). Questi neuroni VTA innervano anche molte altre regioni del proencefalo, tra cui ippocampo, amigdala e corteccia prefrontale (PFC).
Ha senso considerare questi meccanismi di dipendenza da droghe in questo volume in memoria per tre motivi sovrapposti.6
- Innanzitutto, tutti gli adattamenti indotti da farmaci possono essere visti come tipi di "memoria molecolare o cellulare:" le cellule nervose sottoposte a tali cambiamenti sono diverse a causa dell'esposizione al farmaco e quindi reagiscono in modo diverso a quello stesso farmaco, ad altri farmaci, o ad una serie di altri stimoli come risultato.
- In secondo luogo, è interessante notare che molti, forse la maggior parte, dei tipi di cambiamenti che sono stati associati a uno stato di dipendenza (ad es. trascrizione genica alterata, epigenetica, plasticità sinaptica e di cellule intere e morfologia neuronale e meccanismi neurotrofici) sono anche implicati nelle forme tradizionali di "memoria comportamentale" come la memoria spaziale, il condizionamento della paura e il condizionamento operante, tra gli altri.
- Terzo, tra le regioni cerebrali affette da droghe d'abuso ci sono quelle che sono i principali substrati neurali per la memoria comportamentale, inclusi ippocampo, amigdala e PFC. Questo coincide con la crescente consapevolezza che alcune delle caratteristiche più importanti della dipendenza vista clinicamente (ad esempio, il desiderio di droga e la ricaduta) riflettono anomalie nei circuiti di memoria tradizionali, con ricordi a lungo termine dell'esperienza della droga che fungono da potenti motori di patologie da dipendenza.4,7,8 Al contrario, le regioni di ricompensa del cervello (p. Es., VTA e NAc) influenzano potentemente la memoria comportamentale.
Questo articolo fornisce una panoramica dei principali tipi di cambiamenti molecolari e cellulari che si verificano in diverse regioni del cervello in modelli animali di dipendenza, concentrandosi sul nucleo accumbens per il quale è attualmente disponibile la maggior parte delle informazioni. È importante sottolineare che è stato sempre più possibile convalidare alcuni di questi cambiamenti nei tossicodipendenti umani basati su studi sui cervelli postmortem. Nonostante il fatto che le droghe d'abuso abbiano strutture chimiche distinte e agiscano su distinti bersagli proteici, è sorprendente che molti importanti adattamenti legati alla dipendenza siano comuni a molti, e in alcuni casi a tutti, droghe d'abuso e contribuiscano probabilmente a caratteristiche condivise di un sindrome da dipendenza.4,9 Al contrario, molti altri adattamenti indotti da farmaci sono specifici per un dato farmaco e possono mediare aspetti più unici di una data dipendenza. Ci concentriamo qui su droghe d'abuso stimolanti e oppiacee, che producono effetti più drammatici nei modelli animali rispetto ad altri farmaci. Evidenziamo anche aree importanti per future ricerche che aumenteranno ulteriormente la nostra conoscenza delle sindromi da dipendenza e tradurranno questi progressi in migliori test diagnostici e trattamenti.
Meccanismi trascrizionali ed epigenetici
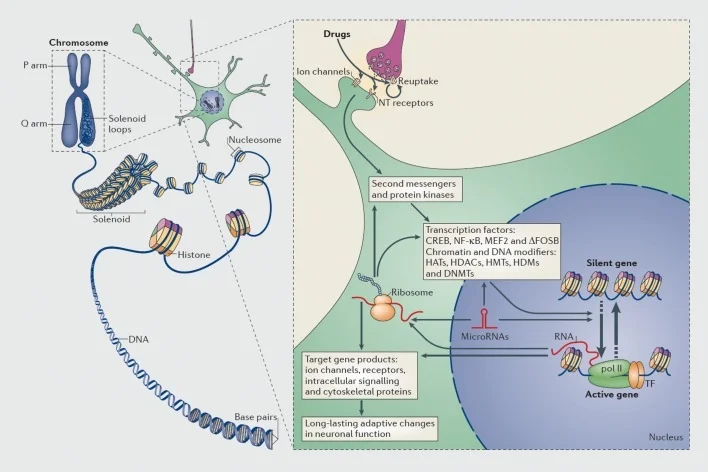
La consapevolezza che i tossicodipendenti possono rimanere ad aumentato rischio di recidiva nonostante anni di astinenza significa che la dipendenza comporta cambiamenti farmacologici nel cervello che possono essere molto stabili. Ciò ha portato diversi gruppi a considerare i cambiamenti nell'espressione genica come una componente importante del processo di dipendenza (Figure 1 ). Di conseguenza, studi di geni candidati o indagini su tutto il genoma che coinvolgono DNA microarrays e più recentemente RNA-seq (sequenziamento ad alto throughput di RNA espressi) ha identificato numerosi geni la cui espressione è alterata in una data regione del cervello nei modelli di dipendenza da roditori e primati e nei tossicodipendenti umani (ad esempio, ref 10-17). Esempi di tali geni sono discussi nelle sezioni successive di questa recensione.
Meccanismi di regolazione trascrizionale ed epigenetica mediante droghe d'abuso. Nelle cellule eucariotiche, il DNA è organizzato avvolgendosi attorno agli ottoni dell'astone per formare nucleosomi, che sono poi ulteriormente organizzati e condensati per formare cromosomi (parte sinistra). Solo distruggendo temporaneamente la cromatina compatta il DNA di un gene specifico può essere reso accessibile al macchinario trascrizionale. Le droghe d'abuso agiscono attraverso bersagli sinaptici come i meccanismi di reuptake, i canali ionici e i recettori del neurotrasmettitore (NT) per alterare le cascate di segnalazione intracellulare (parte destra). Ciò porta all'attivazione o inibizione dei fattori di trascrizione (TF) e di molti altri bersagli nucleari, comprese le proteine regolatrici della cromatina (mostrate da frecce spesse); i meccanismi dettagliati coinvolti nella regolazione sinaptica delle proteine regolatrici della cromatina rimangono scarsamente compresi. Questi processi alla fine portano all'induzione o alla repressione di particolari geni, compresi quelli per RNA non codificanti come i microRNA; l'espressione alterata di alcuni di questi geni può a sua volta regolare ulteriormente la trascrizione genica. Si propone che alcuni di questi cambiamenti indotti dal farmaco a livello della cromatina siano estremamente stabili e quindi alla base dei comportamenti di lunga durata che definiscono la dipendenza. CREB, proteina legante reattiva agli AMP ciclico; DNMT, DNA metiltransferasi; CAPPELLI, istone acetiltransferasi; HDAC, deacetilasi istoniche; HDM, demistasi istoniche; HMT, istone metiltransferasi; MEF2, fattore di potenziamento specifico del miocita 2; NF-kB, fattore-KB nucleare; pol II, RNA polimerasi II. Riprodotto da ref 44: Robison AJ, Nestler EJ. Meccanismi trascrizionali ed epigenetici della dipendenza. Nat Rev Neurosci. 2011; 12: 623-637.
Allo stesso modo, molti tipi di fattori di trascrizione - proteine che si legano alle regioni regolatorie dei geni e quindi aumentano o diminuiscono la trascrizione di quei geni - sono stati implicati nel mediare gli effetti a lungo termine della droga di abuso sull'espressione genica nel cervello. Esempi prominenti includono CREB (proteina di legame dell'elemento di risposta cAMP), ΔFosB (fattore di trascrizione della famiglia Fos), NFkB (fattore nucleare kB), MEF2 (fattore di aumento dei miociti-2) e recettori glucocorticoidi, tra molti altri.5,10,18-22 È stato sempre più possibile comprendere le vie di segnalazione cellulare attraverso le quali le droghe d'abuso attivano un dato fattore di trascrizione nel cervello e collegare causalmente tale attivazione ai geni bersaglio di quel fattore di trascrizione e a specifici aspetti comportamentali della dipendenza (vedere Figura 1). Questo progresso è illustrato dalla considerazione di CREB e ΔFosB, che sono i fattori di trascrizione meglio studiati nei modelli di dipendenza.
proteina di legame dell'elemento di risposta cAMP
Attivano droghe stimolanti e oppiacee CREB in diverse regioni del cervello importanti per la dipendenza, tra cui prominente nel NAc.23,24 È noto che CREB è attivato in altri sistemi da cAMP, Ca2+e percorsi del fattore di crescita,25 e non è ancora noto quale di questi media la sua attivazione in NAc da droghe d'abuso. L'attivazione farmacologica di CREB in NAc ha dimostrato di rappresentare un classico meccanismo di feedback negativo, per cui CREB serve a ridurre la sensibilità di un animale agli effetti gratificanti di questi farmaci (tolleranza) e a mediare uno stato emotivo negativo durante l'astinenza dal farmaco (dipendenza).18,26,27 Questi effetti sono stati recentemente dimostrati per stimolare l'aumento dell'autosomministrazione e della ricaduta della droga, presumibilmente attraverso un processo di rinforzo negativo.28 Queste azioni di CREB sembrano coinvolgere entrambi i principali sottotipi di neuroni spinosi medi di NAc, quelli che esprimono prevalentemente D1 contro D2 recettori della dopamina.24 Inettamente, un ampio corpo di letteratura ha dimostrato che CREB, che agisce nell'ippocampo e nell'amigdala, è una molecola chiave nella memoria comportamentale.29-31 Questo ampio ruolo nella dipendenza e nella memoria comportamentale probabilmente riflette il fatto che i neuroni sono imbevuti di un numero finito di meccanismi molecolari con cui adattarsi ad un ambiente in continua evoluzione.
I geni target per CREB che mediano questo fenotipo comportamentale sono stati identificati attraverso test genome-wide così come più sforzi selezionati.10,18,32 Un esempio è la dinorfina del peptide oppioide: induzione stimolante dell'espressione di dinorfina nei neuroni NAc, mediata tramite CREB, aumenta l'attivazione di dinorfina di k recettori oppioidi sui neuroni della dopamina VTA e quindi sopprime la trasmissione dopaminergica al NAc e danneggia la ricompensa.18 Molti altri bersagli CREB hanno dimostrato di essere importanti per la plasticità sinaptica indotta da farmaci, come discusso di seguito. Mentre CREB è anche attivato in molte altre regioni del cervello da stimolanti e oppiacei,23,24 meno si sa sulle conseguenze comportamentali di questo effetto e sui geni bersaglio attraverso i quali si verificano. Allo stesso modo, si sa meno sul ruolo di CREB nel mediare le azioni di altre droghe d'abuso.19
ΔFosB
L'esposizione acuta a praticamente qualsiasi droga di abuso induce tutti i fattori di trascrizione della famiglia Fos in NAc e in molte altre regioni del cervello. Questa induzione è rapida ma anche altamente transitoria, con livelli di proteina Fos che ritornano alla normalità entro 8 a 12 ore. Unicamente tra queste proteine della famiglia Fos è ΔFosB, un prodotto troncato del gene FosB, che in virtù della sua insolita stabilità, si accumula gradualmente attraverso un ciclo di esposizione ripetuta al farmaco e diventa la proteina Fos predominante espressa in queste condizioni.22,33 Inoltre, a causa di questa stabilità, i livelli di ΔFosB persistono per settimane dopo l'interruzione del farmaco. Tale induzione cronica di ΔFosB è stata dimostrata praticamente per tutte le droghe d'abuso34 e, per la maggior parte dei farmaci, è selettivo per i neuroni NAc di tipo Dl.34,35 È stato anche dimostrato nei dipendenti umani.35 Un ampio corpo di letteratura ha dimostrato che tale induzione ΔFosB in D1-i neuroni di tipo NAc aumentano la sensibilità di un animale ai farmaci così come le ricompense naturali e promuove l'auto-somministrazione del farmaco, presumibilmente attraverso un processo di rinforzo positivo (vedi i riferimenti 34 a 38). È interessante notare che l'induzione della droga di ΔFosB in NAc è più drammatica negli animali adolescenti, un periodo di maggiore vulnerabilità alla dipendenza,39 e la sua induzione da parte della nicotina ha dimostrato di mediare il miglioramento della ricompensa della cocaina simile a un gateway.40
Per quanto riguarda CREB, numerosi geni target per ΔFosB sono stati identificati in NAc mediante l'uso di geni candidato e approcci a livello genomico.10,32 Mentre CREB induce la dinorfina, ΔFosB la sopprime, il che contribuisce agli effetti pro-ricompensa di ΔFosB.38 Un altro obiettivo ΔFosB è cFos: poiché ΔFosB si accumula con l'esposizione ripetuta al farmaco, reprime i c-Fos e contribuisce all'interruttore molecolare con cui ΔFosB viene indotto selettivamente nello stato di terapia farmacologica cronica.41 È stato dimostrato che molti altri bersagli ΔFosB mediano la capacità di alcuni farmaci di abuso di indurre plasticità sinaptica nel NAc e cambiamenti associati nell'arborizzazione dendritica dei neuroni medi spinosi di NAc, come verrà discusso di seguito.
Le conseguenze funzionali dell'induzione di ΔFosB in altre regioni del cervello sono meno conosciute, sebbene la sua induzione nella corteccia orbitofrontale (OFC) sia stata studiata in dettaglio. Qui, ΔFosB media la tolleranza che si verifica agli effetti di distruzione cognitiva della cocaina durante un ciclo di esposizione cronica e questo adattamento è associato ad un aumento dell'autosomministrazione di cocaina.42,43
Saggi a livello genomico hanno suggerito diversi potenziali geni bersaglio che mediano questi effetti.42 Nonostante le proprietà temporali uniche di ΔFosB e la consapevolezza che è indotto nei circuiti di memoria tradizionali (ad esempio, ippocampo), non è stata ancora esplorata il ruolo di ΔFosB nella memoria comportamentale, un argomento interessante per la ricerca futura.
Meccanismi epigenetici
In anni più recenti, gli studi di trascrizione sono stati spinti un passo avanti verso l'epigenetica44 (Vedi Figura 1), che può essere definito in generale come un cambiamento nell'espressione genica che si verifica in assenza di un cambiamento nella sequenza del DNA. I meccanismi epigenetici controllano il confezionamento del DNA all'interno del nucleo di una cellula attraverso le sue interazioni con gli istoni e molti altri tipi di proteine nucleari, che insieme costituiscono la cromatina. L'espressione genica è controllata dallo stato di questo imballaggio attraverso la modifica covalente di istoni, altre proteine e DNA stesso. Come alcuni esempi, l'acetilazione degli istoni tende a promuovere l'attivazione dei geni, la metilazione degli istoni può promuovere l'attivazione o la repressione genica a seconda del residuo di Lys sottoposto a questa modifica e la metilazione del DNA è generalmente associata alla repressione genica, sebbene alcune forme varianti di metilazione ( ad es. 5-idrossimetilazione) può essere associato all'attivazione genica.
L'epigenetica è un meccanismo attraente perché, in altri sistemi, ad esempio la biologia dello sviluppo e del cancro, alcune modificazioni epigenetiche possono essere permanenti. Per questa ragione, l'epigenetica è stata perseguita sia nei modelli di apprendimento e memoria (ad esempio, refs 45-48) che nella dipendenza;44,49 in entrambi i sistemi sono stati riportati profondi cambiamenti nell'acetilazione dell'istone e nella metilazione e nella metilazione del DNA. Come solo un esempio, l'istone metiltransferasi, G9a, è implicata in entrambe le memorie50 e dipendenza51,52 Nei modelli di dipendenza, L'espressione G9a è sottoregolata in NAc in risposta a droghe stimolanti o oppiacee di abuso e til suo ha dimostrato di migliorare gli effetti gratificanti di questi farmaci.51,52 È interessante notare che la soppressione della cocaina di G9a è mediata da ΔFosB. G9a catalizza la dimetilazione di Lys9 dell'istone H3 (H3K9me2), un importante mediatore della repressione genetica. ChIP-chip o ChIP-seq (immunoprecipitazione della cromatina seguita, rispettivamente, da chip promotore o sequenziamento high-throughput) sono stati utilizzati per ottenere mappe genome-wide dei geni in NAc che mostrano alterato H3K9me2 dopo esposizione a stimolante o oppiacei.32,52,53 Sovrapponendo questi elenchi di geni con liste genome-wide di cambiamenti di espressione genica e con mappe a livello genomico di molte altre forme di modificazioni epigenetiche (ad es., Associazione ΔFosB, legame CREB, altre modifiche dell'istone, ecc.),32,53 dovrebbe essere possibile identificare un insieme di geni sempre più completo regolato da droghe d'abuso e comprendere i meccanismi epigenetici sottostanti coinvolti.
Un'altra forma di regolazione epigenetica implicata nella memoria e nella dipendenza è la generazione di microRNA. Questi piccoli RNA non codificanti si legano a regioni complementari di mRNA e quindi sopprimono la loro traduzione o ne inducono la degradazione. La delezione di Argonauta, una proteina cruciale per l'elaborazione dei miRNA, altera le risposte comportamentali alla cocaina, con effetti distinti osservati per i neuroni spinosi medi di tipo D1- contro D2.54 Parecchi miRNA specifici hanno anche dimostrato di essere regolati dall'esposizione ai farmaci e, a loro volta, di influenzare le risposte comportamentali ai farmaci (ad esempio, i ref 55,56). Sarà interessante negli studi futuri identificare gli obiettivi degli mRNA di questi miRNA e caratterizzare il modo in cui influenzano il processo di dipendenza.
Plasticità sinaptica
Gli stessi tipi generali di modificazioni sinaptiche alle sinapsi glutammatergiche, che sono state implicate nell'ippocampo e nell'amigdala nella memoria comportamentale (vedi altri articoli in questo numero), hanno anche dimostrato di verificarsi nelle regioni di ricompensa cerebrale nei modelli di dipendenza e di essere importanti nella mediazione il processo di dipendenza.57,58 Tale plasticità sinaptica indotta da farmaci è stata descritta in diverse regioni del cervello, tuttavia, ci concentriamo qui su NAc, dove la maggior parte della ricerca si è concentrata fino ad oggi (Figure 2 ).
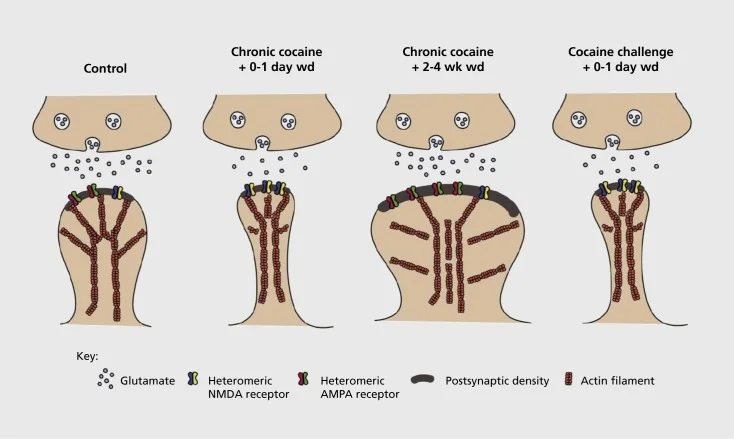
Modello di plasticità sinaptica e strutturale legata alla dipendenza nel nucleo accumbens (NAc). L'esposizione cronica alla cocaina determina una riorganizzazione transitoria e temporanea di recettori di glutammato di acido α-ammino-3-idrossi-5-metil-4-isossazolapropionico (AMPA) e N-metil-D-aspartico (NMDA) di glutammato in terreno NAc sinapsi del neurone spinoso (MSN), così come i cambiamenti strutturali nella testa della colonna vertebrale degli MSN di NAc che si correlano con forme distinte di plasticità sinaptica. Ad esempio, la cocaina cronica induce l'espressione superficiale dei recettori NMDA, la formazione silenziosa delle sinapsi e la depressione a lungo termine (LTD) nei punti di tempo di sospensione precoce. Durante il ritiro più prolungato (wd), queste alterazioni sinaptiche si invertono con il risultato di una maggiore espressione dei recettori AMPA di superficie, un consolidamento della sinapsi in una spina a forma di fungo e un potenziamento a lungo termine (LTP). Questi effetti tornano rapidamente indietro dopo un'esposizione a una dose di sfida di cocaina che porta alla ristrutturazione della spina dorsale in sottili spine e una depressione della forza sinaptica.
Gli esperimenti iniziali hanno dimostrato che l'esposizione ripetuta a droghe stimolanti di abuso induce uno stato simile a una depressione (a lungo termine) a sinapsi glutammatergiche nel NAc.59 Tuttavia, un lavoro più recente ha dimostrato che tale plasticità è altamente dipendente dal tempo, con la comparsa di LTD subito dopo l'ultima esposizione alla cocaina che si evolve in più di uno stato LTP (potenziamento a lungo termine) dopo periodi di sospensione più lunghi.60,61 Questo lavoro, che fino ad oggi è stato condotto principalmente utilizzando uno sperimentatore somministrato - al contrario dei farmaci auto-somministrati, ha definito la necessità di indagini più sistematiche nei modelli di autoamministrazione che tracciano le forme di plasticità sinaptica che si verificano nelle sinapsi glutamatergiche in NAc su un corso temporale dettagliato dall'acquisizione di autoamministrazione al suo mantenimento, attraverso diversi tempi di ritiro ed estinzione, e in risposta a stimoli che richiamano la ricaduta. I lavori fino ad oggi hanno anche definito alcuni dei meccanismi molecolari che contribuiscono a questa plasticità sinaptica indotta da farmaci, incluso il traffico di recettori AMPA alla sinapsi forse mediata in parte tramite CaMKII (Ca2+/ chinasi della proteina-calmodulina-dipendente II) fosforilazione di alcune subunità del recettore AMPA nonché espressione alterata delle subunità del recettore AMPA (es. 60,62-65, Figure 2 e 3). Un ruolo per CREB e ΔFosB è stato implicato in questi fenomeni, così come nei cambiamenti associati nella morfologia delle sinapsi glutamatergiche (vedi sotto). Ad esempio, GluAl è un bersaglio per CREB in NAc, dove GluA2 e CaMKII sono entrambi bersagli di ΔFosB, in questa regione del cervello .35,36,66,67 Andando avanti, sarà importante collegare specifici adattamenti ai cambiamenti dipendenti dal tempo nella funzione sinaptica e le caratteristiche comportamentali della dipendenza.

Meccanismi molecolari alla base dell'induzione da cocaina di spine dendritiche sui neuroni spinosi medi del nucleo accumbens (NAc). A) mostra aumenti indotti dalla cocaina nel numero della colonna vertebrale dendritica che possono essere bloccati dalla sovraespressione virale di G9a o JunD (un antagonista della trascrizione mediata da AP1) o imitati dalla sovraespressione virale di FosB. B) È stato dimostrato che la regolazione del traffico del recettore AMPA (AMPAR) e del citoscheletro di actina (a sinistra), nonché la regolazione della trascrizione dei recettori del glutammato e delle proteine regolatrici dell'actina (p. Es., Mediate tramite ΔFosB, a destra) svolgono un ruolo importante nel mediare la regolazione della cocaina della densità della colonna vertebrale dendritica NAc. UMK, chinasi del dominio LIM; RAC, substrato della tossina botulinica C3 correlata a Ras.
Nuovi strumenti sperimentali stanno rendendo possibile per la prima volta definire con crescente precisione quali particolari circuiti mostrano queste forme di plasticità sinaptica e quali anomalie comportamentali mediano. Ad esempio, tLe subregioni di shell e core di NAc mostrano differenze nella plasticità sinaptica indotta da farmaci, così come D1- rispetto ai neuroni spinosi medi di tipo D2 all'interno di ciascuna subregione.60,63,64,67 Allo stesso modo, gli esperimenti optogenetici hanno fornito nuove informazioni sul contributo di una particolare forma di plasticità sinaptica (ad es., LTD) a specifiche popolazioni di sinapsi glutammatergiche in NAc, ad esempio quelle derivanti dalla PFC mediale rispetto all'amigdala basolaterale rispetto al subiculum ventrale (la principale produzione di ippocampo).68-70 In definitiva, sarà necessario sovrapporre adattamenti molecolari indotti da farmaci in ciascuno di questi neuroni afferenti con adattamenti sinapsi specifici che si verificano nei loro dendriti postsinaptici per compilare una comprensione completa di come le droghe d'abuso modificano i circuiti del cervello per guidare particolari aspetti del stato dipendente. Questo sforzo richiederà un maggiore apprezzamento della plasticità indotta dai farmaci nelle sinapsi inibitorie all'interno di queste stesse regioni del cervello, un'area che ha ricevuto pochissima attenzione fino ad oggi.65
Plasticità di cellule intere
Mentre la maggior parte della ricerca che coinvolge i cambiamenti neurofisiologici nei neuroni nei fenomeni di abuso di droghe, come nei fenomeni di apprendimento e memoria, si è concentrata sulla plasticità sinaptica, ci sono sempre più prove per l'importanza della plasticità delle cellule intere. Plasticità delle cellule intere, anche indicata come plasticità omeostatica,71 comporta cambiamenti nell'eccitabilità intrinseca di un'intera cellula nervosa in un modo che non è specifico della sinapsi. Dato che alcune caratteristiche della tossicodipendenza implicano una sensibilità aumentata o ridotta a un farmaco, ha senso che l'eccitabilità elettrica potenziata o ridotta di alcune cellule nervose contribuisca a questi adattamenti comportamentali.5
I il miglior esempio di plasticità delle cellule intere a un farmaco di abuso è la capacità degli oppiacei cronici di aumentare l'eccitabilità intrinseca dei neuroni noradrenergici del locus coeruleus (LC).72 Questa aumentata eccitabilità è mediata dal CREB e dalla sua induzione di alcune isoforme di adenilil ciclasi, che determinano un aumento del rilascio dei neuroni LC, forse attraverso l'induzione dei canali Na +.72-75 Questa ipereccitazione dei neuroni LC rappresenta un meccanismo classico di tolleranza e dipendenza e guida alcuni dei segni e sintomi dell'astinenza da oppiacei. È interessante notare che CREB media una simile forma di plasticità delle cellule intere nei neuroni medi spinosi NAc, che sono anche resi ipereccitabili dall'esposizione cronica a droghe d'abuso tramite CREB.76 Sarà quindi fondamentale nelle indagini future capire come la plasticità sinaptica CREB-mediata delle sinapsi glutammatergiche sui neuroni medi spinosi NAc65,66 riassume con ipereccitabilità intrinseca mediata da CREB di questi neuroni76 controllare le caratteristiche comportamentali della dipendenza.
Un altro esempio di plasticità delle cellule intere nei modelli di dipendenza è l'ipereccitabilità dei neuroni della dopamina VTA che si verifica dopo l'esposizione cronica a farmaci oppiacei di abusoe (Figure 4 ).77,78 Questo adattamento, che è stato collegato ai cambiamenti morfologici in queste cellule nervose (si veda la prossima sezione), non è mediato da CREB ma ottenuto attraverso la regolazione delle cascate di segnalazione neurotrofica, come descritto di seguito.
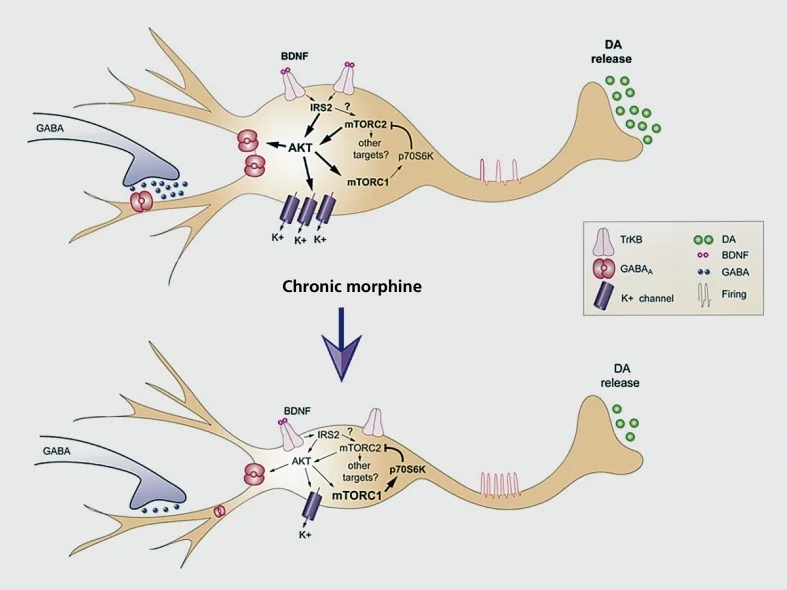
Modello funzionante di adattamenti cronici indotti dalla morfina nei neuroni della dopamina con area tegmentale ventrale (VTA). La morfina cronica diminuisce la dimensione del soma della dopamina (DA) VTA ma aumenta l'eccitabilità neuronale, mentre la trasmissione della dopamina al nucleo accumbens è diminuita. L'effetto netto della morfina è una via di ricompensa meno reattiva, cioè la tolleranza alla ricompensa. Downregulation di IRS2-AKT segnalazione in VTA media gli effetti della morfina cronica sulla dimensione del soma e l'eccitabilità elettrica; l'effetto sull'eccitabilità è mediato dalla diminuzione delle correnti di acido γ-amminobutirrico (GABA) A e dalla soppressione dell'espressione del canale K '. La downregolazione indotta dalla morfina dell'attività di mTORC2 in VTA è cruciale per questi adattamenti morfologici e fisiologici indotti dalla morfina, nonché per la tolleranza alla ricompensa. In contrasto con mT0RC2, la morfina cronica aumenta l'attività mTORCI, che non influenza questi adattamenti indotti dalla morfina. BDNF, fattore neurotrofico derivato dal cervello; IRS, sostanza del recettore dell'insulina; mTORC, complesso mTOR; AKT, protein chinasi B Riprodotto da ref 77
Plasticità morfologica e meccanismi neurotrofici
L'aumento delle prove, in gran parte da studi su neuroni corticali ippocampali e cerebrali, ha dimostrato che i cambiamenti nella plasticità sinaptica sono associati a cambiamenti morfologici alle sinapsi. Ad esempio, LTD e la generazione di sinapsi silenti sono associate alla formazione di spine dendritiche sottili o tozze, mentre LTP è associato a spine più grandi a forma di fungo.79,80 È quindi interessante che il campo dell'abuso di droghe si sia concentrato sui cambiamenti indotti da farmaci nelle spine dendritiche per> 15 anni. Esposizione cronica a droghe stimolanti di abuso aumenta la densità della colonna vertebrale dendritica dei neuroni spinosi medi del NAc, un cambiamento che predomina per i neuroni del tipo Dl.67,81,82 L'induzione delle spine è stata associata per la maggior parte a risposte comportamentali sensibilizzate a questi farmaci, sebbene alcune prove siano in conflitto con questa visione.
Come per gli studi sulla plasticità sinaptica, tuttavia, è necessario molto più lavoro per definire sistematicamente i cambiamenti nelle spine dendritiche che si verificano durante un ciclo di auto-somministrazione di farmaci, astinenza e recidiva. Sle ricerche finora svolte, che coinvolgono farmaci sperimentali e auto-somministrati, suggeriscono cambiamenti della colonna vertebrale molto diversi che si verificano in diversi punti temporali di astinenza e in una shell NAc rispetto alle subregioni principali.83-86 Sarà anche importante definire i meccanismi molecolari precisi con cui la cocaina o un altro stimolante produce effetti specifici dipendenti dal tempo e dal tipo di cellula. ΔFosB ha dimostrato di essere sia necessario che sufficiente per l'induzione di spine immature su neuroni NAc di tipo Dl.35,51,67 Tale regolazione avviene di concerto con la cocaina e la regolazione ΔFosB di diverse proteine note per controllare la riorganizzazione del citoscheletro di actina. Come solo un esempio, la regolazione trascrizionale di diversi fattori di scambio nucleotidico guanina e proteine attivanti GTPase mette in relazione Rac1, una piccola GTPasi, per diminuzioni transitorie dell'attività in risposta a ciascuna esposizione di cocaina e tali diminuzioni pulsatili nell'attività di Rac1 sono state dimostrate utilizzando il controllo optogenetico di Rac1, per mediare l'induzione di spine immature.87 Questi effetti di Racl presumibilmente si verificano attraverso il suo controllo di cofilin e altre proteine regolatrici dell'actina, che hanno anche dimostrato di mediare la regolazione della cocaina della crescita della colonna vertebrale.87,88 Tuttavia, è importante sottolineare che questo è solo un percorso coinvolto nella regolazione della cocaina delle spine acerbe, poiché è stato dimostrato che anche molte altre proteine svolgono un ruolo essenziale, tra cui CDK5 (chinasi dipendente dalla ciclina-5), CaMKII, NFkB , MEF2, CREB, G9a e DNMT3 (DNA methyltransf cancella 3a), solo per citarne alcuni.20,21,35,51,67,89,90 È interessante notare che la regolazione della cocaina di molti di questi geni, inclusa l'induzione di CDK5, CaMKII e NFkB e la repressione di G9a, è anche mediata da ΔFosB.20,35,51,91
Sorprendentemente, le droghe di abuso di oppiacei esercitano l'effetto opposto e riducono la densità della colonna vertebrale dendritica dei neuroni spinosi medi di NAc.81 Poco si sa delle conseguenze comportamentali di questo adattamento e dei meccanismi molecolari sottostanti coinvolti. Questo fenomeno è, tuttavia, sorprendente, dato che CREB e ΔFosB sono indotti sia da stimolanti che da oppiacei e sono entrambi implicati nell'induzione mediata dallo stimolo della densità della colonna dendritica di NAc. Ciò solleva la questione di come gli oppiacei sopprimono la densità della colonna vertebrale NAc nonostante la loro induzione di questi fattori.
L'altra importante forma di plasticità morfologica osservata nei modelli di abuso di droghe è la riduzione fisica delle dimensioni del soma cellulare dei neuroni della dopamina VTA indotta dalla somministrazione cronica di oppiacei.77,92,93 Un adattamento simile si verifica in risposta ai cannabinoidi.94 Questo restringimento dei neuroni della dopamina VTA, che si verifica con l'auto-somministrazione degli oppiacei93 ed è stato documentato in eroina umana esaminata postmortem,77 sembra mediare la tolleranza della ricompensa ed è associato con un ridotto rilascio di dopamina nel NAc. Prove considerevoli ora indicano che questa riduzione delle dimensioni del soma cellulare è mediata dalla soppressione degli oppiacei dell'espressione del fattore neurotrofico derivato dal cervello (BDNF) all'interno di questi neuroni. Abbiamo direttamente collegato questa sospensione indotta da oppiacei del supporto BDNF e la contrazione del neurone VTA, a ridotta attività delle cascate di segnalazione BDNF a valle nei neuroni della dopamina VTA, specificamente ridotta attività di IRS2 (substrato del recettore insulinico-2), AKT (una serina-treonina chinasi) e TORC2 (bersaglio della rapamicina-2, che è insensibile alla rapamicina).77,93 Abbiamo anche collegato questa downregulation della segnalazione di BDNF direttamente all'aumentata eccitabilità che la morfina induce in questi neuroni, come notato in precedenza.77,78 In effetti, le dimensioni ridotte del soma cellulare e l'aumentata eccitabilità sono strettamente accoppiate, poiché l'induzione dell'uno conduce all'altra e viceversa. Questo controllo sull'eccitabilità cellulare implica la soppressione di K+ canali e di GABAA corrente in questi neuroni.
Questo ruolo del BDNF nel controllo delle risposte alla morfina a livello del VTA contrasta con il suo coinvolgimento molto diverso nelle azioni della cocaina e di altri stimolanti. Gli stimolanti inducono la segnalazione del BDNF al NAc, un effetto dovuto all'aumentata sintesi locale del BDNF e un aumento del rilascio da diverse regioni afferenti.95 Inoltre, l'aumento della segnalazione BDNF in NAc, ma non nel VTA, ha dimostrato di promuovere gli effetti comportamentali di questi farmaci inclusa la loro auto-somministrazione.95,96 La regolazione opposta della segnalazione di BDNF nella via VTA-NAc da parte degli oppiacei rispetto agli stimolanti solleva la possibilità che tali differenze mediano la regolazione opposta dei farmaci delle spine dendritiche NAc, una possibilità ora sotto studio.
Direzioni future
La suddetta narrazione sottolinea gli enormi progressi che sono stati fatti nella comprensione degli adattamenti molecolari e cellulari che si verificano nelle regioni di ricompensa del cervello in risposta all'esposizione ripetuta a una droga di abuso e nel correlare adattamenti individuali a certe caratteristiche comportamentali delle sindromi da dipendenza in modelli animali . Nonostante questi progressi, restano importanti domande. La maggior parte delle nostre conoscenze attuali si concentra su VTA e NAc, con molte meno informazioni disponibili su altre regioni chiave del cervello limbico che sono anche cruciali per la tossicodipendenza. Inoltre, tutte le dimostrazioni sperimentali del ruolo causale di un adattamento molecolare-cellulare in un comportamento correlato alla droga hanno manipolato adattamenti individuali uno alla volta. La manipolazione di numerosi adattamenti contemporaneamente è chiaramente molto più difficile, ma è anche essenziale, poiché sappiamo che i farmaci producono un gran numero di diversi tipi di cambiamenti anche all'interno dei singoli neuroni, il che probabilmente riassume in modi complicati l'influenza del comportamento. Un tale approccio alla biologia dei sistemi sarà fondamentale per spezzare in definitiva le basi biologiche della dipendenza. Infine, gli sforzi per comprendere i meccanismi molecolari-cellulari dei ricordi legati alla dipendenza si trovano nel punto in cui tutti gli altri sforzi per comprendere le basi biologiche della memoria comportamentale ora combattono: la nostra capacità di mettere in relazione i fenomeni biologici con la memoria comportamentale complessa rimane estremamente difficile. Superare questa divisione rappresenta forse la più grande sfida nelle neuroscienze.
Ringraziamenti
Questo lavoro è stato supportato da sovvenzioni dall'istituto nazionale per l'abuso di droghe.
Abbreviazioni e acronimi selezionati
- Nac
- nucleo accumbens
- CREB
- proteina di legame dell'elemento di risposta del cAMP
- ΔFosB
- un fattore di trascrizione della famiglia Fos
- VTA
- area tegmentale ventrale
- AMPA
- acido α-ammino-3-idrossi-5-metil-4-isossazolapropionico
- LTD
- depressione a lungo termine
- LTP
- protenrio a lungo termine
- BDNF
- fattore neurotrofico derivato dal cervello
- NKkB
- fattore nucleare kB
BIBLIOGRAFIA