COMENTÁRIOS: Como estudos posteriores irão revelar, DeltaFosB é o interruptor molecular comum para drogas e vícios comportamentais. É um fator de transcrição, o que significa que afeta quais genes são ativados ou desativados. Como afirmado em outro lugar, as drogas aditivas apenas sequestram os mecanismos normais. É por isso que é tolo sugerir que os vícios comportamentais não podem existir.
Proc Natl Acad Sci EUA A. 2001 setembro 25; 98 (20): 11042 – 11046.
doi: 10.1073 / pnas.191352698.
Eric J. Nestler *, Michel Barrot e David W. Self
Departamento de Psiquiatria e Centro de Neurociência Básica, Centro Médico Southwestern da Universidade do Texas, 5323 Harry Hines Boulevard, Dallas, TX 75390-9070
Sumário
A longevidade de algumas das anormalidades comportamentais que caracterizam a dependência de drogas tem sugerido que a regulação da expressão gênica neural pode estar envolvida no processo pelo qual as drogas de abuso causam um estado de dependência. EuEvidências crescentes sugerem que o fator de transcrição ΔFosB representa um mecanismo pelo qual as drogas de abuso produzem mudanças relativamente estáveis no cérebro que contribuem para o fenótipo de dependência. ΔFosB, um membro da família Fos de fatores de transcrição, se acumula dentro de um subgrupo de neurônios do nucleus accumbens e estriado dorsal (regiões do cérebro importantes para dependência) após a administração repetida de muitos tipos de drogas de abuso. Acumulação semelhante de ΔFosB ocorre após a corrida compulsiva, o que sugere que ΔFosB pode se acumular em resposta a muitos tipos de comportamentos compulsivos. É importante ressaltar que ΔFosB persiste nos neurônios por períodos de tempo relativamente longos devido à sua extraordinária estabilidade. Portanto, ΔFosB representa um mecanismo molecular que poderia iniciar e sustentar mudanças na expressão gênica que persistem por muito tempo após a exposição à droga cessar.. Estudos em camundongos transgênicos induzíveis que superexpressam ΔFosB ou um inibidor negativo dominante da proteína fornecem evidências diretas de que ΔFosB causa aumento da sensibilidade aos efeitos comportamentais de drogas de abuso e, possivelmente, aumento do comportamento de busca por drogas. Este trabalho sustenta a visão de que ΔFosB funciona como um tipo de “troca molecular” sustentada que gradualmente converte respostas agudas a drogas em adaptações relativamente estáveis que contribuem para a plasticidade neural e comportamental de longo prazo subjacente ao vício.
A pesquisa sobre vícios está focada na compreensão das maneiras complexas pelas quais as drogas de abuso alteram o cérebro para causar anormalidades comportamentais que caracterizam o vício. Um dos desafios críticos no campo é identificar mudanças relativamente estáveis induzidas por drogas no cérebro para explicar as anormalidades comportamentais que são particularmente duradouras. Por exemplo, um adicto humano pode estar em maior risco de recaída, mesmo após anos de abstinência.
A estabilidade dessas anormalidades comportamentais levou à sugestão de que elas podem ser mediadas, pelo menos em parte, por mudanças na expressão gênica (1-3). De acordo com esse ponto de vista, a exposição repetida a uma droga de abuso perturba repetidamente a transmissão em determinadas sinapses no cérebro que são sensíveis à droga. Tais perturbações eventualmente sinalizam através de cascatas de mensageiros intracelulares para o núcleo, onde elas primeiro iniciam e depois mantêm mudanças na expressão de genes específicos. Um mecanismo primário pelo qual as vias de transdução de sinal influenciam a expressão gênica é a regulação de fatores de transcrição, proteínas que se ligam a regiões reguladoras de genes e modificam sua transcrição.
Um objetivo da pesquisa de dependência, portanto, foi identificar fatores de transcrição que são alterados em regiões do cérebro implicadas na dependência após administração crônica de drogas de abuso. Vários desses fatores de transcrição foram identificados na última década (1-6). O foco desta revisão está em um fator de transcrição particular chamado ΔFosB.
Indução de ΔFosB por drogas de abuso
ΔFosB, codificado pelo gene fosB, é um membro da família Fos dos fatores de transcrição, que também inclui c-Fos, FosB, Fra1 e Fra2 (7). Estas proteínas da família Fos heterodimerizam com proteínas da família Jun (c-Jun, JunB ou JunD) para formar fatores ativos de transcrição AP-1 (proteína ativadora-1) que se ligam a sítios AP-1 (seqüência de consenso: TGAC / GTCA) os promotores de certos genes para regular sua transcrição.
Estas proteínas da família Fos são induzidas rápida e transitoriamente em regiões cerebrais específicas após a administração aguda de muitas drogas de abuso (Fig. 1) (8-11). As regiões proeminentes são o nucleus accumbens e o estriado dorsal, que são importantes mediadores das respostas comportamentais às drogas, em particular, seus efeitos recompensadores e de ativação locomotora (12, 13). Essas proteínas retornam aos níveis basais em poucas horas após a administração da droga.
Figura 1
Esquema mostrando a acumulação gradual de ΔFosB versus a indução rápida e transitória de outras proteínas da família Fos em resposta a drogas de abuso. (A) O autorradiograma ilustra a indução diferencial dessas várias proteínas por estimulação aguda (1-2 h após exposição a um único medicamento) versus estimulação crônica (1 dia após a exposição repetida ao medicamento). (B) Várias ondas de proteínas semelhantes a Fos [compreendidas por c-Fos (isoformas 52- a 58-kDa), FosB (isoformas 46- a 50-kDa), ΔFosB (isoforma 33-kDa) e Fra1 ou Fra2 ( 40 kDa)] são induzidos no núcleo accumbens e nos neurônios do estriado dorsal pela administração aguda de uma droga de abuso. Também induzidas são isoformas modificadas bioquimicamente de ΔFosB (35-37 kDa); eles também são induzidos (embora em níveis baixos) após a administração aguda de drogas, mas persistem no cérebro por longos períodos por causa de sua estabilidade. (C) Com a administração repetida (por exemplo, duas vezes ao dia) de medicamentos, cada estímulo agudo induz um baixo nível das isoformas ΔFosB estáveis, o que é indicado pelo menor conjunto de linhas sobrepostas que indicam ΔFosB induzido por cada estímulo agudo. O resultado é um aumento gradual nos níveis totais de ΔFosB com estímulos repetidos durante um curso de tratamento crônico, o que é indicado pela crescente linha escalonada no gráfico.
Respostas muito diferentes são observadas após a administração crônica de drogas de abuso (Fig. 1). As isoformas de ΔFosB modificadas bioquimicamente (massa molecular 35-37 kDa) acumulam-se nas mesmas regiões do cérebro após a exposição repetida ao fármaco, enquanto todos os outros membros da família Fos mostram tolerância (isto é, redução da indução comparada com exposições iniciais ao fármaco). Tal acumulação de ΔFosB foi observada para cocaína, morfina, anfetamina, álcool, nicotina e fenciclidinae (11, 14 – 18). Há alguma evidência de que essa indução é seletiva para o subconjunto de dinossauro espinhoso contendo dinorfina / substância localizado nessas regiões do cérebro (15, 17), embora seja necessário mais trabalho para estabelecer isso com certeza. As isoformas 35- a 37-kDa de ΔFosB dimerizam predominantemente com JunD para formar um complexo AP-1 ativo e de longa duração nestas regiões cerebrais (19, 20). Estas isoformas de ΔFosB se acumulam com a exposição crônica a medicamentos devido a suas meias-vidas extraordinariamente longas (21) e, portanto, persistem nos neurônios por pelo menos várias semanas após a cessação da administração da droga. É interessante notar que estas isoformas ΔFosB são produtos altamente estáveis de um gene precoce imediato (fosB). A estabilidade das isoformas de ΔFosB fornece um novo mecanismo molecular pelo qual as alterações induzidas pela droga na expressão gênica podem persistir, apesar de períodos relativamente longos de retirada da droga.
Embora o núcleo accumbens desempenhe um papel crítico nos efeitos recompensadores das drogas de abuso, acredita-se que ele funcione normalmente regulando as respostas aos reforçadores naturais, como alimentos, bebidas, sexo e interações sociais (12, 13). Como resultado, há um interesse considerável em um possível papel dessa região cerebral em outros comportamentos compulsivos (por exemplo, excessos patológicos, jogos de azar, exercícios, etc.). Por essa razão, examinamos se ΔFosB é regulado em um modelo animal de corrida compulsiva. De fato, as isoformas estáveis 35- a 37-kDa de ΔFosB são induzidas seletivamente dentro do nucleus accumbens em ratos que apresentam comportamento de corrida compulsiva. †
Identidade Bioquímica de Isoformas Estáveis ΔFosB
Como mencionado acima, as isoformas de ΔFosB que se acumulam após a administração crónica de uma droga de abuso ou corrida compulsiva mostram uma massa molecular de 35-37 kDa. Eles podem ser diferenciados da isoforma 33-kDa de ΔFosB que é induzida rapidamente, mas transitoriamente, após uma única exposição a fármaco (Fig. 1) (14, 19, 22). Evidências atuais sugerem que a isoforma 33-kDa é a forma nativa da proteína, que é alterada para formar os produtos mais estáveis 35- a 37-kDa (19, 21). No entanto, a natureza da modificação bioquímica que converte a isoforma instável 33-kDa nas isoformas estáveis 35- a 37-kDa permaneceu obscura. Tem sido especulado que a fosforilação pode ser responsável (11). Por exemplo, a indução de ΔFosB é atenuada em ratos sem DARPP-32, uma proteína enriquecida no estriado (23, 24). Como o DARPP-32 regula a atividade catalítica da proteína fosfatase-1 e da proteína quinase A (25, 26), a exigência dessa proteína para o acúmulo normal das isoformas estáveis de ΔFosB sugere um possível papel para a fosforilação na geração desses produtos estáveis.
Papel de ΔFosB em Plasticidade Comportamental para Drogas de Abuso
O insight sobre o papel de ΔFosB na dependência de drogas veio em grande parte do estudo de camundongos transgênicos nos quais ΔFosB pode ser induzido seletivamente dentro do núcleo accumbens e outras regiões estriatais de animais adultos (27, 28). É importante ressaltar que esses camundongos superexpressam ΔFosB seletivamente nos neurônios espinhosos mediados pela dinorfina / substância P, onde se acredita que os fármacos induziriam a proteína. O fenótipo comportamental dos camundongos que superexpressam ΔFosB, que em muitos aspectos se assemelham a animais após exposição crônica a medicamentos, está resumido na Tabela 1. Os ratos mostram respostas locomotoras aumentadas à cocaína após administração aguda e crônica (28). Eles também mostram maior sensibilidade aos efeitos recompensadores da cocaína e da morfina nos ensaios de condicionamento local (11, 28) e auto-administrarão doses menores de cocaína do que irmãos que não expressam em excesso ΔFosB. ‡ Em contraste, esses animais apresentam locomotores condicionados normais. sensibilização à cocaína e aprendizagem espacial normal no labirinto aquático de Morris (28). TEsses dados indicam que ΔFosB aumenta a sensibilidade de um animal à cocaína e talvez a outras drogas de abuso e pode representar um mecanismo para uma sensibilização relativamente prolongada às drogas.
 striatum
striatum
| Aumento da ativação locomotora em resposta à administração aguda e repetida de cocaína. |
| Respostas de recompensa aumentadas à cocaína e morfina em ensaios de condicionamento de lugar. |
| Aumento da auto-administração de baixas doses de cocaína. |
| Maior motivação para cocaína em ensaios de proporção progressiva. |
| Respostas ansiolíticas aumentadas ao álcool. |
| Aumento do comportamento de corrida compulsiva. |
Baseado em dados em refs. 28 e 29.† ‡ §¶
Plasticidade comportamental mediada por ΔFosB no núcleo accumbens-striatum dorsal
IAlém disso, há evidências preliminares de que os efeitos da ΔFosB podem se estender muito além de uma regulação da sensibilidade da droga per se a comportamentos mais complexos relacionados ao processo de dependência. Camundongos expressando ΔFosB trabalham mais para auto-administrar cocaína em ensaios de auto-administração de razão progressiva, sua ingestão de ΔFosB pode sensibilizar os animais para as propriedades motivacionais de incentivo da cocaína e, assim, levar a uma propensão para a recaída após a retirada da drogaOs camundongos com expressão ‡ osFosB também mostram efeitos ansiolíticos aumentados do álcool, § um fenótipo que tem sido associado ao aumento da ingestão de álcool em humanos. Juntas, essas descobertas iniciais sugerem que ΔFosB, além de aumentar a sensibilidade a drogas de abuso, produz mudanças qualitativas no comportamento que promovem o comportamento de busca de drogas. Assim, ΔFosB pode funcionar como um “interruptor molecular” sustentado que ajuda a iniciar e manter aspectos cruciais do estado viciado. Uma questão importante sob investigação atual é se o acúmulo de ΔFosB durante a exposição ao medicamento promove o comportamento de busca de drogas após períodos prolongados de retirada, mesmo após os níveis de ΔFosB terem se normalizado (veja abaixo).
Adulto camundongos que superexpressam ΔFosB seletivamente dentro do nucleus accumbens e estriado dorsal também exibem maior corrida compulsiva comparados com ninhadas de controle. † Essas observações levantam a possibilidade interessante de que a acumulação de ΔFosB dentro desses neurônios atua de forma mais geral na formação e manutenção de memórias de hábitos e compulsão. comportamentos, talvez reforçando a eficácia dos circuitos neurais nos quais esses neurônios atuam.
ΔFosB acumula-se em certas regiões do cérebro fora do núcleo accumbens e estriado dorsal após exposição crônica à cocaína. Proeminente entre estes regiões são a amígdala e córtex pré-frontal medial (15) Um dos principais objetivos da pesquisa atual é entender as contribuições da indução de ΔFosB nessas regiões para o fenótipo de dependência.
Trabalhos anteriores em camundongos knockout para fosB revelaram que esses animais não conseguem desenvolver sensibilização aos efeitos locomotores da cocaína, o que é consistente com os achados dos camundongos com superexpressão de ΔFosB mencionados acima (22). No entanto, os mutantes fosB mostraram maior sensibilidade aos efeitos agudos da cocaína, o que é inconsistente com esses outros achados. A interpretação dos resultados com os mutantes fosB, no entanto, é complicada pelo fato de que esses animais não têm apenas ΔFosB, mas também FosB de comprimento total. Além disso, os mutantes carecem de proteínas em todo o cérebro e desde os primeiros estágios de desenvolvimento. Na verdade, um trabalho mais recente apóia as conclusões dos camundongos com superexpressão de ΔFosB: a superexpressão induzível de um mutante truncado de c-Jun, que atua como um antagonista negativo dominante de ΔFosB, seletivamente no nucleus accumbens e estriado dorsal mostra sensibilidade reduzida aos efeitos recompensadores da cocaína .¶ Essas descobertas enfatizam o cuidado que deve ser usado na interpretação dos resultados de camundongos com mutações constitutivas e ilustram a importância dos camundongos com mutações induzíveis e específicas do tipo de célula em estudos de plasticidade no cérebro adulto.
Genes alvo para ΔFosB
Como ΔFosB é um fator de transcrição, presumivelmente, a proteína causa plasticidade comportamental por meio de alterações na expressão de outros genes. ΔFosB é gerado por splicing alternativo do gene fosB e não possui uma porção do domínio de transativação C-terminal presente em FosB de comprimento total. Como resultado, foi originalmente proposto que ΔFosB funciona como um repressor de transcrição (29). No entanto, o trabalho em cultura de células demonstrou claramente que ΔFosB pode induzir ou reprimir Transcrição mediada por AP-1 dependendo do local AP-1 usado (21, 29-31). O FosB de comprimento total exerce os mesmos efeitos que o ΔFosB em certos fragmentos do promotor, mas efeitos diferentes em outros. Mais trabalho é necessário para entender os mecanismos subjacentes a essas ações variadas de ΔFosB e FosB.
Nosso grupo usou duas abordagens para identificar genes-alvo para ΔFosB. Uma é a abordagem do gene candidato. Inicialmente, consideramos os receptores de glutamato do ácido α-amino-3-hidroxi-5-metil-4-isoxazolpropiônico (AMPA) como alvos putativos, dado o importante papel da transmissão glutamatérgica no nucleus accumbens. O trabalho até o momento indicou que uma subunidade particular do receptor de glutamato AMPA, GluR2, pode ser um alvo fidedigno para ΔFosB (Fig. 2). A expressão de GluR2, mas não a expressão de outras subunidades do receptor AMPA, é aumentada no nucleus accumbens (mas não no estriado dorsal) após a superexpressão de ΔFosB (28), e a expressão de um mutante negativo dominante atenua a capacidade da cocaína de induzir a proteína.¶ Além disso, o promotor do gene GluR2 contém um sítio AP-1 de consenso que se liga a ΔFosB (28). A superexpressão de GluR2 no nucleus accumbens, pelo uso de transferência gênica mediada por vírus, aumenta a sensibilidade de um animal aos efeitos recompensadores da cocaína, imitando assim parte do fenótipo visto nos camundongos que expressam ΔFosB (28). A indução de GluR2 poderia ser responsável pela sensibilidade eletrofisiológica reduzida dos neurônios do nucleus accumbens aos agonistas do receptor AMPA após a administração crônica de cocaína (32), porque os receptores AMPA contendo GluR2 mostram condutância geral reduzida e permeabilidade ao Ca2 + reduzida. A resposta reduzida desses neurônios a estímulos excitatórios pode, então, aumentar as respostas a uma droga de abuso. No entanto, as formas pelas quais os sinais dopaminérgicos e glutamatérgicos no nucleus accumbens regulam o comportamento aditivo permanecem desconhecidas; isso exigirá um nível de compreensão do circuito neural, que ainda não está disponível.
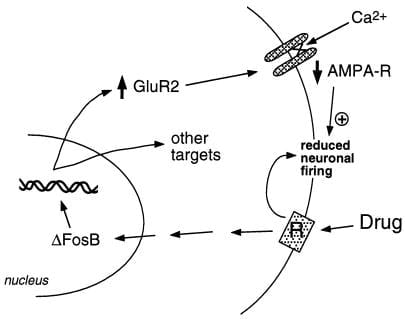
Figura 2
A subunidade do receptor de glutamato AMPA, GluR2, é um alvo putativo para ΔFosB. É mostrado como a indução de GluR2 mediada por ΔFosB pode alterar a responsividade fisiológica dos neurônios do nucleus accumbens e levar a respostas sensibilizadas a drogas de abuso. De acordo com esse esquema, as drogas de abuso produzem seus efeitos de reforço agudos por meio da inibição dos neurônios do núcleo accumbens. Com a exposição repetida, as drogas induzem ΔFosB, que regula vários genes-alvo, incluindo GluR2. Isso aumenta a proporção de receptores AMPA (AMPA-R) nos neurônios do núcleo accumbens que contêm a subunidade GluR2, o que causa redução da corrente AMPA geral e redução da corrente de Ca2 +. Essa excitabilidade reduzida pode tornar os neurônios mais sensíveis aos efeitos inibitórios agudos das drogas e, portanto, aos efeitos de reforço das drogas.
Outro alvo putativo para ΔFosB é o gene que codifica a dinorfina. Como afirmado anteriormente, a dinorfina é expressa no subgrupo de neurônios espinhosos médios do nucleus accumbens que mostram a indução de ΔFosB. A dinorfina parece funcionar em um circuito de retroalimentação intercelular: sua liberação inibe os neurônios dopaminérgicos que inervam os neurônios espinhosos médios, via receptores opioides κ presentes nas terminações nervosas dopaminérgicas no núcleo accumbens e também nos corpos celulares e dendritos na região tegmentar ventral. (Fig. 3) (33 – 35). Esta ideia é consistente com a capacidade de um agonista do receptor κ, após a administração em qualquer uma destas duas regiões do cérebro, para diminuir od (35).
RUm trabalho eficiente indicou que ΔFosB diminui a expressão de dinorfina, ‖ o que poderia contribuir para o aumento dos mecanismos de recompensa vistos com a indução ΔFosB. Curiosamente, outro fator de transcrição regulado por drogas, CREB (proteína de ligação ao elemento de resposta cAMP) (2, 3), exerce o efeito oposto: induz a expressão da dinorfina no nucleus accumbens e reduz as propriedades recompensadoras da cocaína e da morfina (4). **
BA ativação induzida pelo fármaco do CREB dissipa-se rapidamente após a administração do fármaco, tal regulação recíproca da dinorfina pelo CREB e ΔFosB poderia explicar as alterações comportamentais recíprocas que ocorrem durante as fases precoces e tardias de abstinência, com sintomas emocionais negativos e sensibilidade reduzida ao fármaco predominando nas fases iniciais de retirada e sensibilização para os efeitos motivacionais de recompensa e incentivo de drogas que predominam em momentos posteriores.
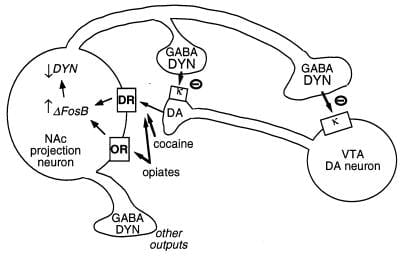
Figura 3
A dinorfina é um alvo putativo para ΔFosB. É mostrado um neurônio de dopamina (DA) de área tegmentar ventral (VTA) que inerva uma classe de neurônios de projeção GABAérgica do núcleo accumbens (NAc) que expressa dinorfina (DYN). A dinorfina serve como um mecanismo de feedback neste circuito: a dinorfina, liberada dos terminais dos neurônios NAc, atua nos receptores opioides κ localizados nas terminações nervosas e nos corpos celulares dos neurônios DA para inibir seu funcionamento. ΔFosBinibindo a expressão de dinorfina, pode regular negativamente este ciclo de realimentação e melhorar as propriedades recompensadoras de drogas de abuso. Não mostrado é o efeito recíproco da CREB neste sistema: CREB aumenta a expressão de dinorfina e, portanto, atenua as propriedades recompensadoras de drogas de abuso (4) GABA, ido? -Aminobutico; DR, receptor de dopamina; OU, receptor opióide.
A segunda abordagem usada para identificar genes alvo para ΔFosB envolve a análise de microarranjos de DNA. A superexpressão induzível de ΔFosB aumenta ou diminui a expressão de vários genes no nucleus accumbens (36). Embora um trabalho considerável seja necessário agora para validar cada um desses genes como alvos fisiológicos de ΔFosB e para entender sua contribuição para o fenótipo de dependência, um alvo importante parece ser Cdk5 (cinase-5 dependente de ciclina). Assim, o Cdk5 foi inicialmente identificado como ΔFosB-regulado pelo uso de microarrays, e mais tarde mostrou ser induzido no nucleus accumbens e estriado dorsal após administração crônica de cocaína (37). ΔFosB ativa o gene cdk5 através de um local AP-1 presente no promotor do gene (36). Juntos, esses dados suportam um esquema em que a cocaína induz a expressão de Cdk5 nessas regiões do cérebro por meio de ΔFosB. A indução de Cdk5 parece alterar a sinalização dopaminérgica, pelo menos em parte, através do aumento da fosforilação de DARPP-32 (37), que é convertido de um inibidor da proteína fosfatase-1 em um inibidor da proteína quinase A mediante sua fosforilação por Cdk5 (26).
Papel de ΔFosB na mediação da plasticidade "permanente" às drogas de abuso
Embora o sinal ΔFosB seja relativamente duradouro, ele não é permanente. ΔFosB degrada gradualmente e não pode mais ser detectado no cérebro após meses de abstinência de drogas, mesmo que certas anormalidades comportamentais persistam por períodos muito mais longos. Portanto, ΔFosB por si só não parece ser capaz de mediar essas anormalidades comportamentais semipermanentes. A dificuldade em encontrar as adaptações moleculares subjacentes às mudanças comportamentais extremamente estáveis associadas ao vício é análoga aos desafios enfrentados no campo da aprendizagem e da memória. Embora existam modelos celulares e moleculares elegantes de aprendizado e memória, até hoje não foi possível identificar adaptações moleculares e celulares suficientemente duradouras para explicar memórias comportamentais altamente estáveis. Na verdade, ΔFosB é a adaptação de vida mais longa conhecida por ocorrer no cérebro adulto, não apenas em resposta a drogas de abuso, mas também a qualquer outra perturbação (que não envolva lesões). Duas propostas surgiram, tanto no campo do vício quanto no campo do aprendizado e da memória, para dar conta dessa discrepância.
Uma possibilidade é que mudanças mais transitórias na expressão gênica, como aquelas mediadas via ΔFosB ou outros fatores de transcrição (por exemplo, CREB), pode mediar mudanças mais duradouras na morfologia neuronal e na estrutura sináptica. Por exemplo, nos um aumento na densidade das espinhas dendríticas (particularmente um aumento nas espinhas de duas cabeças) aumento da eficácia das sinapses glutamatérgicas em neurônios piramidais do hipocampo durante a potenciação de longo prazo (38-40), e paralela a maior sensibilidade comportamental à cocaína mediada no nível dos neurônios espinhosos médios do nucleus accumbens (41). Não se sabe se tais mudanças estruturais são suficientemente duradouras para levar em conta mudanças altamente estáveis no comportamento, embora estas persistam por pelo menos o mês de retirada da droga. Evidências recentes levantam a possibilidade de que ΔFosB, e sua indução de Cdk1, seja um mediador de alterações induzidas por drogas na estrutura sináptica no nucleus accumbens (Fig. 5). ‡ Assim, a infusão de um inibidor de Cdk4 no nucleus accumbens impede a capacidade de exposição repetida à cocaína para aumentar a densidade da coluna dendrítica nesta região. Isso é consistente com a visão de que o Cdk5, que é enriquecido no cérebro, regula a estrutura neural e o crescimento (ver ref. 5 e 36). É possível, embora de maneira alguma provado, que tais mudanças na morfologia neuronal possam durar mais que o próprio sinal de ΔFosB.
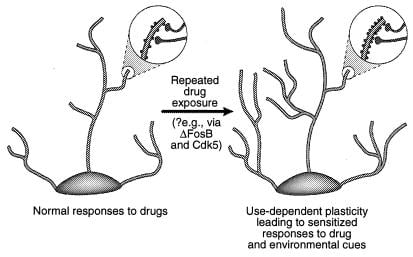
Figura 4
Regulação da estrutura dendrítica por drogas de abuso. É mostrada a expansão da árvore dendrítica de um neurônio após exposição crônica a uma droga de abuso, como foi observado com a cocaína no nucleus accumbens e no córtex pré-frontal (41). As áreas de ampliação mostram um aumento nas espinhas dendríticas, que é postulado como ocorrendo em conjunto com terminais nervosos ativados. Este aumento na densidade da coluna dendrítica pode ser mediado por ΔFosB e a consequente indução de Cdk5 (ver texto). Essas alterações na estrutura dendrítica, que são semelhantes às observadas em alguns modelos de aprendizagem (por exemplo, potencialização de longo prazo), podem mediar respostas sensibilizadas de longa duração a drogas de abuso ou estímulos ambientais. [Reproduzido com permissão da ref. 3 (Copyright 2001, Macmillian Magazines Ltd.)].
Outra possibilidade é que a indução transitória de um fator de transcrição (por exemplo, ΔFosB, CREB) leva a mudanças mais permanentes na expressão gênica através da modificação de cromatosn. Crê-se que estes e muitos outros factores de transcrição activam ou reprimem a transcrição de um gene alvo, promovendo a acetilação ou desacetilação, respectivamente, de histonas na vizinhança do gene (42). Embora tal acetilação e desacetilação de histonas possam aparentemente ocorrer muito rapidamente, é possível que ΔFosB ou CREB possam produzir adaptações mais duradouras na maquinaria enzimática que controla a acetilação de histonas. ΔFosB ou CREB também podem promover mudanças mais duradouras na expressão gênica, regulando outras modificações da cromatina (por exemplo, metilação de DNA ou histona) que foram implicadas nas mudanças permanentes na transcrição gênica que ocorrem durante o desenvolvimento (ver ref. 42 e 43) . Embora essas possibilidades permaneçam especulativas, elas poderiam fornecer um mecanismo pelo qual adaptações transitórias a uma droga de abuso (ou alguma outra perturbação) levam a conseqüências comportamentais essencialmente vitalícias.
Referências
- ↵
- Nestler EJ,
- Espero BT,
- Widnell KL
(1993) Neurônio 11: 995 – 1006.
CrossRefMedlineSite: da Ciência
- ↵
- Berke JD,
- Hyman SE
(2000) Neurônio 25: 515 – 532.
CrossRefMedlineSite: da Ciência
- ↵
- Nestler EJ
(2001) Nat Rev Neurosci 2: 119-128.
CrossRefMedlineSite: da Ciência
- ↵
- Carlezon WA Jr,
- Thome J,
- Olson VG,
- Lane-Ladd SB,
- Brodkin ES,
- Hiroi N,
- Duman RS,
- Neve RL,
- Nestler EJ
(1998) Ciência 282: 2272 – 2275.
-
- O'Donovan KJ,
- Tourtellotte WG,
- Millbrandt J,
- Baraban JM
(1999) Tendências Neurosci 22: 167 – 173.
- ↵
- Mackler SA,
- Korutla L,
- Cha XY,
- Koebbe MJ,
- Fournier KM,
- Bowers MS,
- Kalivas PW
(2000) J Neurosci 20: 6210-6217.
- ↵
- Morgan JI,
- Curran T
(1995) Tendências Neurosci 18: 66 – 67.
- ↵
- ST jovem
- Porrino LJ,
- Iadarola MJ
(1991) Proc Natl Acad Sci EUA 88: 1291 – 1295.
-
- Graybiel AM,
- Moratalla R,
- Robertson HA
(1990) Proc Natl Acad Sci EUA 87: 6912 – 6916.
-
- Esperança B,
- Kosofsky B,
- Hyman SE,
- Nestler EJ
(1992) Proc Natl Acad Sci EUA 89: 5764 – 5768.
- ↵
- Kelz MB,
- Nestler EJ
(2000) Curr Opin Neurol 13: 715 - 720.
CrossRefMedlineSite: da Ciência
- ↵
- Koob GF,
- Sanna PP,
- Bloom FE
(1998) Neurônio 21: 467 – 476.
CrossRefMedlineSite: da Ciência
- ↵
- RA sábio
(1998) Dependência de álcool de drogas 51: 13 – 22.
CrossRefMedlineSite: da Ciência
- ↵
- Espero BT,
- Nye HE,
- Kelz MB,
- Auto DW,
- Iadarola MJ,
- Nakabeppu Y,
- Duman RS,
- Nestler EJ
(1994) Neurônio 13: 1235 – 1244.
- ↵
- Nye H,
- Espero BT,
- Kelz M,
- Iadarola M,
- Nestler EJ
(1995) J Pharmacol Exp Ther 275: 1671-1680.
-
- Nye HE,
- Nestler EJ
(1996) Mol Pharmacol 49: 636-645.
- ↵
- Moratalla R,
- Elibol B,
- Vallejo M,
- Graybiel AM
(1996) Neurônio 17: 147 – 156.
CrossRefMedlineSite: da Ciência
- ↵
- Pich EM,
- Pagliusi SR,
- Tessari M,
- Talabot-Ayer D,
- Hooft van Huijsduijnen R,
- Chiamulera C
(1997) Ciência 275: 83 – 86.
- ↵
- Chen JS,
- Nye HE,
- Kelz MB,
- Hiroi N,
- Nakabeppu Y,
- Espero BT,
- Nestler EJ
(1995) Mol Pharmacol 48: 880-889.
- ↵
- Hiroi N,
- Marrom J,
- Ye H,
- Saudou F,
- Vaidya VA,
- Duman RS,
- Greenberg ME,
- Nestler EJ
(1998) J Neurosci 18: 6952-6962.
- ↵
- Chen J,
- Kelz MB,
- Espero BT,
- Nakabeppu Y,
- Nestler EJ
(1997) J Neurosci 17: 4933-4941.
- ↵
- Hiroi N,
- Marrom J,
- Haile C,
- Ye H,
- Greenberg ME,
- Nestler EJ
(1997) Proc Natl Acad Sci EUA 94: 10397 – 10402.
- ↵
- Fienberg AA,
- Hiroi N,
- Mermelstein P,
- Canção WJ,
- Snyder GL,
- Nishi A,
- Cheramy A,
- O'Callaghan JP,
- Miller D,
- Cole DG,
- et ai.
(1998) Ciência 281: 838 – 842.
- ↵
- Hiroi N,
- Feinberg A,
- Haile C,
- Greengard P,
- Nestler EJ
(1999) Eur J Neurosci 11: 1114-1118.
CrossRefMedlineSite: da Ciência
- ↵
- Greengard P,
- Allen PB,
- Nairn AC
(1999) Neurônio 23: 435 – 447.
CrossRefMedlineSite: da Ciência
- ↵
- Bibb JA,
- Snyder GL,
- Nishi A,
- Yan Z,
- Meijer L,
- Fienberg AA,
- Tsai LH,
- Kwon YT,
- Girault JA,
- Czernik AJ,
- et ai.
(1999) Nature (Londres) 402: 669 – 671.
- ↵
- Chen JS,
- Kelz MB,
- Zeng GQ,
- Sakai N,
- Steffen C,
- Shockett PE,
- Picciotto M,
- Duman RS,
- Nestler EJ
(1998) Mol Pharmacol 54: 495-503.
- ↵
- Kelz MB,
- Chen JS,
- Carlezon WA,
- Whisler K,
- Gilden L,
- Beckmann AM,
- Steffen C,
- Zhang YJ,
- Marotti L,
- Auto SW,
- et ai.
(1999) Nature (Londres) 401: 272 – 276.
- ↵
- Dobrazanski P,
- Noguchi T,
- Kovary K,
- Rizzo CA,
- Lazo PS,
- Bravo R
(1991) Mol Cell Biol 11: 5470-5478.
-
- Nakabeppu Y,
- Nathans D
(1991) Cell 64: 751 – 759.
CrossRefMedlineSite: da Ciência
- ↵
- Yen J,
- Sabedoria RM,
- Tratner I,
- Verma IM
(1991) Proc Natl Acad Sci EUA 88: 5077 – 5081.
- ↵
- FJ branco
- Hu XT,
- Zhang XF,
- Wolf ME
(1995) J Pharmacol Exp Ther 273: 445-454.
- ↵
- Hyman SE
(1996) Neurônio 16: 901 – 904.
-
- Kreek MJ
(1997) Pharmacol Biochem Behav 57: 551-569.
CrossRefMedlineSite: da Ciência
- ↵
- TS de Shippenberg,
- Rea W
(1997) Pharmacol Biochem Behav 57: 449-455.
CrossRefMedlineSite: da Ciência
- ↵
- Chen JS,
- Zhang YJ,
- Kelz MB,
- Steffen C,
- Ang ES,
- Zeng L,
- Nestler EJ
(2000) J Neurosci 20: 8965-8971.
- ↵
- Bibb JA,
- Chen JS,
- Taylor JR,
- Svenningsson P,
- Nishi A,
- Snyder GL,
- Yan Z,
- Sagawa ZK,
- Nairn AC,
- Nestler EJ,
- et ai.
(2001) Nature (Londres) 410: 376 – 380.
- ↵
- Luscher C,
- Nicoll RA,
- Malenka RC,
- Muller D
(2000) Nat Neurosci 3: 545-550.
CrossRefMedlineSite: da Ciência
-
- Malinow R,
- Mainen ZF,
- Hayashi Y
(2000) Curr Opin Neurobiol 10: 352-357.
CrossRefMedlineSite: da Ciência
- ↵
- Scannevin RH,
- Huganir RL
(2000) Nat Rev Neurosci 1: 133-141.
CrossRefMedlineSite: da Ciência
Robinson, TE & Kolb, B. (1999) (1997) EUR. J. Neurosci.11, 1598-1604.
- ↵
- Carey M,
- Smale ST
(2000) Regulação transcricional em eucariotos (Cold Spring Harbor Lab. Press, Plainview, NY).
- ↵
- Spencer VA,
- Davie JR
(1999) Gene 240: 1 – 12.
CrossRefMedlineSite: da Ciência
 Facebook
Facebook Twitter
Twitter- Google+
 CiteULike
CiteULike Delicious
Delicious Digg
Digg Mendeley
Mendeley
Artigos hospedados pela imprensa HighWire citando este artigo
- As recompensas naturais e de drogas atuam em mecanismos comuns de plasticidade neural com {Delta} FosB como um mediador chave J. Neurosci. 2013 33 (8) 3434-3442
- Drogas, Crime e Epigenética da Alostase Hedônica Revista de Justiça Criminal Contemporânea 2012 28 (3) 314-328
- Sumário
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- Sumário
- Texto Completo (HTML)
- Texto Completo (PDF)
- A Morfina Ativa o Fator de Transcrição E vinte e seis como-1 / Via do Fator de Resposta para Soro via Quinases Reguladas por Sinal Extracelular 1 / 2 em Células F11 Derivadas de Neurônios dos Gânglios da Raiz Dorsal J. Pharmacol. Exp. Ther. 2012 342 (1) 41-52
- Mecanismo Molecular para uma Droga de Entrevista: Mudanças Epigenéticas Iniciadas pela Nicotina Expressão de Gene Principal pela Cocaína Sci Transl Med 2011 3 (107) 107ra109
- Autoadministração Aprimorada de Sacarose e Cocaína e Busca de Drogas Induzidas por Cue após a Perda de VGLUT2 em Neurônios Dopaminérgicos Mesencéfalos em Ratos J. Neurosci. 2011 31 (35) 12593-12603
- Hipóxia intermitente crônica aumenta pressão sanguínea e expressão de FosB / {Delta} FosB em regiões autonômicas centrais Sou. J. Physiol. Regul. Integrar Comp. Physiol. 2011 301 (1) R131-R139
- Ausência do receptor GPR37 / PAEL prejudica a fosforilação estriatal de Akt e ERK2, expressão de {Delta} FosB e condiciona a preferência de lugar para anfetaminas e cocaína FASEB 2011 25 (6) 2071-2081
- A relação entre a duração da exposição inicial ao álcool e a persistência da tolerância molecular é marcadamente não-linear J. Neurosci. 2011 31 (7) 2436-2446
- Imagem de Bioluminescência In Vivo Revela Ativação Proteína-1 Regulada por Redox no Núcleo Paraventricular de Camundongos com Hipertensão Renovascular Hipertensão 2011 57 (2) 289-297
- A superexpressão do estria de {Delta} FosB reproduz movimentos involuntários induzidos por levodopa crônica J. Neurosci. 2010 30 (21) 7335-7343
- Mediação Epigenética de Influências Ambientais em Distúrbios Psicóticos Maiores Touro Schizophr 2009 35 (6) 1045-1056
- Sondas de ressonância magnética baseadas em DNA para detecção específica de exposição crônica a anfetaminas em cérebros vivos J. Neurosci. 2009 29 (34) 10663-10670
- Plasticidade Alterada da Coluna Dendrítica em Ratos Retirados de Cocaína J. Neurosci. 2009 29 (9) 2876-2884
- A tela de superexpressão em Drosophila identifica os papéis neuronais da GSK-3 {beta} / shaggy como um regulador da plasticidade desenvolvimental dependente de AP-1 Genética 2008 180 (4) 2057-2071
- Transcrição MRI: uma nova visão do cérebro vivo Neurocientista 2008 14 (5) 503-520
- Indução de FosB {Delta} no córtex orbitofrontal medeia a tolerância à disfunção cognitiva induzida por cocaína J. Neurosci. 2007 27 (39) 10497-10507
- Vulnerabilidade permanente para a reintegração do comportamento de busca de metanfetaminas em camundongos mutantes do fator neurotrófico derivados da linhagem de células gliais FASEB 2007 21 (9) 1994-2004
- {Delta} FosB no Núcleo Accumbens regula comportamento e motivação instrumental reforçados com alimentos J. Neurosci. 2006 26 (36) 9196-9204
- Regulação da estabilidade de {Delta} FosB por fosforilação. J. Neurosci. 2006 26 (19) 5131-5142
- Expressão de Receptores Mutantes de NMDA em Células Contendo Receptores de Dopamina D1 Previne Sensibilização da Cocaína e Diminui Preferência de Cocaína J. Neurosci. 2005 25 (28) 6651-6657
- Os Receptores de Dopamina D1 Modulam a Indução FosB {Delta} no Estriato de Ratos após Administração Intermitente de Morfina J. Pharmacol. Exp. Ther. 2005 314 (1) 148-154
- Neurobiologia de camundongos selecionados para alta atividade voluntária de corrida em roda Integrar Comp. Biol. 2005 45 (3) 438-455
- Efeitos da privação de água e reidratação na coloração de c-Fos e FosB no núcleo supraóptico de rato e na região da lâmina terminal Sou. J. Physiol. Regul. Integrar Comp. Physiol. 2005 288 (1) R311-R321
- Indução transcricional do gene FosB / {Delta} FosB por estresse mecânico em osteoblastos J Biol Chem 2004 279 (48) 49795-49803
- Indução de {Delta} FosB em estruturas cerebrais relacionadas a recompensas após estresse crônico J. Neurosci. 2004 24 (47) 10594-10602
- A dosagem do gene Sim1 modula a resposta alimentar homeostática ao aumento da gordura dietética em camundongos Sou. J. Physiol. Endocrinol. Metab. 2004 287 (1) E105-E113
- Análise de DNA microarray da expressão gênica em astrócitos da cabeça do nervo óptico humano em resposta à pressão hidrostática Physiol. Genomics 2004 17 (2) 157-169
- O superóxido está envolvido na ativação do sistema nervoso central e na simpatobicitação da insuficiência cardíaca induzida por infarto do miocárdio Circ. Res. 2004 94 (3) 402-409
- Receptores de adenosina A2A em neuroadaptação à estimulação dopaminérgica repetida: implicações para o tratamento de discinesias na doença de Parkinson Neurologia 2003 61 (90116) S74-81
- Localização Citoplasmática Versus Nuclear de Proteínas Relacionadas a Fos no Sapo, Rana esculenta, Testículos: In Vivo e Efeito In Vitro Direto de um Agonista do Hormônio Liberador de Gonadotropina Biol. Reprod. 2003 68 (3) 954-960
- Ratos Periadolescent mostram aumento da regulação positiva de Delta FosB em resposta à cocaína e à anfetamina J. Neurosci. 2002 22 (21) 9155-9159
- Delta FosB regula o funcionamento das rodas J. Neurosci. 2002 22 (18) 8133-8138
- Atividade de CREB na casca do nucleus accumbens controla o controle das respostas comportamentais aos estímulos emocionais Proc. Natl. Acad. Sci. EUA 2002 99 (17) 11435-11440
- Psicogenômica: Oportunidades para entender o vício J. Neurosci. 2001 21 (21) 8324-8327