KOMMENTARE: Wie spätere Studien zeigen werden, ist DeltaFosB der übliche molekulare Schalter sowohl für Drogen- als auch für Verhaltensabhängigkeiten. Es ist ein Transkriptionsfaktor, der bedeutet, dass er beeinflusst, welche Gene ein- oder ausgeschaltet werden. Wie an anderer Stelle erwähnt, entführen Suchtmittel nur normale Mechanismen. Deshalb ist es albern zu behaupten, dass Verhaltensabhängigkeiten nicht existieren können.
Proc Natl Acad Sci USA A. 2001 September 25; 98 (20): 11042-11046.
doi: 10.1073 / pnas.191352698.
Eric J. Nestler *, Michel Barrot und David W. Self
Abteilung für Psychiatrie und Zentrum für grundlegende Neurowissenschaften, Texas University Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, Texas 75390-9070
Abstract
Die Langlebigkeit einiger der Verhaltensabnormalitäten, die die Drogenabhängigkeit charakterisieren, legt nahe, dass die Regulation der neuralen Genexpression in den Prozess involviert sein könnte, durch den Missbrauchsdrogen einen Suchtstatus verursachen. ichZunehmende Beweise legen nahe, dass der Transkriptionsfaktor ΔFosB einen Mechanismus darstellt, durch den Missbrauchsdrogen relativ stabile Veränderungen im Gehirn hervorrufen, die zum Suchtphänotyp beitragen. ΔFosB, ein Mitglied der Fos - Familie von Transkriptionsfaktoren, akkumuliert in einer Untergruppe von Neuronen des Nucleus accumbens und des dorsalen Striatums (für Sucht relevante Gehirnregionen) nach wiederholter Verabreichung vieler Arten von Missbrauchsdrogen. Eine ähnliche Akkumulation von & Dgr; FosB tritt nach einem zwanghaften Lauf auf, was darauf hindeutet, dass & Dgr; FosB sich als Reaktion auf viele Arten von zwanghaften Verhaltensweisen akkumulieren kann. Wichtig ist, dass ΔFosB aufgrund seiner außerordentlichen Stabilität relativ lange in Neuronen persistiert. Daher stellt ΔFosB einen molekularen Mechanismus dar, der Veränderungen in der Genexpression einleiten und aufrechterhalten kann, die noch lange nach dem Ende der Arzneimittelexposition bestehen bleiben. Studien an induzierbaren transgenen Mäusen, die entweder ΔFosB oder einen dominant negativen Inhibitor des Proteins überexprimieren, liefern den direkten Beweis, dass ΔFosB eine erhöhte Sensitivität für die Verhaltenswirkungen von Missbrauchsdrogen verursacht und möglicherweise ein erhöhtes Drogensuchverhalten. Diese Arbeit unterstützt die Ansicht, dass ΔFosB als eine Art anhaltender "molekularer Schalter" fungiert, der akute Arzneimittelreaktionen allmählich in relativ stabile Anpassungen umwandelt, die zur langfristigen neuralen und Verhaltensplastizität beitragen, die der Sucht zugrunde liegt.
Suchtforschung konzentriert sich auf das Verständnis der komplexen Wege, auf denen Missbrauchsdrogen das Gehirn verändern, um Verhaltensauffälligkeiten zu verursachen, die Sucht charakterisieren. Eine der kritischen Herausforderungen auf diesem Gebiet besteht darin, relativ stabile arzneimittelinduzierte Veränderungen im Gehirn zu identifizieren, um jene Verhaltensauffälligkeiten zu berücksichtigen, die besonders langlebig sind. Zum Beispiel kann ein menschlicher Süchtiger auch nach Jahren der Abstinenz ein erhöhtes Rezidivrisiko haben.
Die Stabilität dieser Verhaltensauffälligkeiten hat zu dem Vorschlag geführt, dass sie zumindest teilweise durch Veränderungen der Genexpression (1-3) vermittelt werden können. Gemäß dieser Ansicht stört die wiederholte Exposition gegenüber einem Missbrauchsdrogen wiederholt die Übertragung an bestimmten Synapsen im Gehirn, die für das Arzneimittel empfindlich sind. Solche Störungen signalisieren schließlich über intrazelluläre Messenger-Kaskaden den Zellkern, wo sie zuerst Veränderungen in der Expression spezifischer Gene initiieren und dann aufrechterhalten. Ein primärer Mechanismus, durch den Signaltransduktionswege die Genexpression beeinflussen, ist die Regulation von Transkriptionsfaktoren, Proteinen, die an regulatorische Regionen von Genen binden und ihre Transkription modifizieren.
Ein Ziel der Suchtforschung war es daher, Transkriptionsfaktoren zu identifizieren, die in Gehirnregionen verändert sind, die nach chronischer Verabreichung von Missbrauchsdrogen an der Sucht beteiligt sind. Mehrere dieser Transkriptionsfaktoren wurden in den letzten zehn Jahren identifiziert (1-6). Der Fokus dieser Überprüfung liegt auf einem bestimmten Transkriptionsfaktor namens ΔFosB.
Induktion von ΔFosB durch Drogen des Missbrauchs
ΔFosB, kodiert durch das fosB-Gen, ist ein Mitglied der Fos-Familie der Transkriptionsfaktoren, zu denen auch c-Fos, FosB, Fra1 und Fra2 (7) gehören. Diese Proteine der Fos-Familie heterodimerisieren mit Proteinen der Jun-Familie (c-Jun, JunB oder JunD), um aktive AP-1 (Aktivator-Protein-1) Transkriptionsfaktoren zu bilden, die an AP-1-Stellen (Konsensussequenz: TGAC / GTCA) binden die Promotoren bestimmter Gene, um ihre Transkription zu regulieren.
Diese Proteine der Fos-Familie werden nach akuter Verabreichung vieler Missbrauchsdrogen in spezifischen Hirnregionen schnell und transient induziert (Abb. 1) (8-11). Prominente Regionen sind der Nucleus accumbens und das dorsale Striatum, die wichtige Mediatoren der Verhaltensreaktionen auf die Medikamente sind, insbesondere ihre lohnenden und bewegungsaktivierenden Wirkungen (12, 13). Diese Proteine kehren innerhalb von Stunden nach der Arzneimittelverabreichung zu den Basalspiegeln zurück.
Figure 1
Schema zeigt die allmähliche Akkumulation von ΔFosB gegen die schnelle und transiente Induktion anderer Proteine der Fos-Familie als Reaktion auf Missbrauchsdrogen. (A) Das Autoradiogramm veranschaulicht die differentielle Induktion dieser verschiedenen Proteine durch akute Stimulation (1-2 hr nach einer einzelnen Arzneimittelexposition) gegenüber chronischer Stimulation (1-Tag nach wiederholter Arzneimittelexposition). (B) Mehrere Wellen von Fos-ähnlichen Proteinen [bestehend aus c-Fos (52- zu 58-kDa-Isoformen), FosB (46- zu 50-kDa-Isoformen), ΔFosB (33-kDa-Isoform) und Fra1 oder Fra2 ( 40 kDa)] werden im Nucleus accumbens und in den dorsalen striatalen Neuronen durch akute Verabreichung eines Missbrauchsdrogens induziert. Ebenfalls induziert werden biochemisch modifizierte Isoformen von ΔFosB (35-37 kDa); sie werden auch nach akuter Arzneimittelverabreichung (wenn auch in geringen Mengen) induziert, bleiben jedoch wegen ihrer Stabilität im Gehirn für lange Zeit bestehen. (C) Bei wiederholter (z. B. zweimal täglicher) Arzneimittelverabreichung induziert jeder akute Stimulus ein niedriges Niveau der stabilen ΔFosB-Isoformen, was durch die untere Reihe von überlappenden Linien angezeigt wird, die durch jeden akuten Stimulus induziertes ΔFosB anzeigen. Das Ergebnis ist ein allmählicher Anstieg der Gesamtspiegel von ΔFosB mit wiederholten Stimuli während eines Verlaufs der chronischen Behandlung, was durch die zunehmende abgestufte Linie in dem Graph angezeigt wird.
Nach chronischer Verabreichung von Drogen sind sehr unterschiedliche Reaktionen zu beobachten (Abb. 1). Biochemisch modifizierte Isoformen von ΔFosB (Molekülmasse 35-37 kDa) akkumulieren nach wiederholter Arzneimittelexposition in den gleichen Hirnregionen, während alle anderen Mitglieder der Fos-Familie eine Toleranz zeigen (dh eine verminderte Induktion im Vergleich zu anfänglichen Arzneimittelexpositionen). Eine solche Akkumulation von ΔFosB wurde für Kokain, Morphin, Amphetamin, Alkohol, Nikotin und Phencyclidin beobachtete (11, 14-18). Es gibt einige Hinweise, dass diese Induktion selektiv für die Dynorphin / Substanz P-haltige Untergruppe von medium-stacheligen Neuronen in diesen Gehirnregionen (15, 17) ist, obwohl mehr Arbeit benötigt wird, um dies mit Sicherheit zu etablieren. Die 35- bis 37-kDa-Isoformen von ΔFosB dimerisieren vorwiegend mit JunD, um einen aktiven und langlebigen AP-1-Komplex innerhalb dieser Gehirnregionen (19, 20) zu bilden. Diese & Dgr; FosB-Isoformen akkumulieren aufgrund ihrer außerordentlich langen Halbwertszeiten (21) bei chronischer Arzneimittelexposition und verbleiben daher in den Neuronen für mindestens einige Wochen nach Beendigung der Arzneimittelverabreichung. Es ist interessant festzustellen, dass diese ΔFosB-Isoformen sehr stabile Produkte eines unmittelbaren frühen Gens (fosB) sind. Die Stabilität der & Dgr; FosB-Isoformen stellt einen neuartigen molekularen Mechanismus bereit, durch den arzneimittelinduzierte Veränderungen in der Genexpression trotz relativ langer Perioden des Arzneimittelabzugs bestehen bleiben können.
Obwohl der Nucleus Accumbens eine entscheidende Rolle bei den belohnenden Wirkungen von Missbrauchsdrogen spielt, wird angenommen, dass er normal funktioniert, indem er Reaktionen auf natürliche Verstärker wie Nahrung, Alkohol, Sex und soziale Interaktionen reguliert (12, 13). Daher besteht ein erhebliches Interesse an einer möglichen Rolle dieser Hirnregion bei anderen zwanghaften Verhaltensweisen (z. B. pathologisches Überessen, Glücksspiel, Sport usw.). Aus diesem Grund haben wir untersucht, ob ΔFosB in einem Tiermodell für zwanghaftes Laufen reguliert wird. Tatsächlich werden die stabilen 35- bis 37-kDa-Isoformen von ΔFosB selektiv im Nucleus Accumbens bei Ratten induziert, die zwanghaftes Laufverhalten zeigen. †
Biochemische Identität von stabilen ΔFosB-Isoformen
Wie oben erwähnt, zeigen die & Dgr; FosB-Isoformen, die sich nach chronischer Verabreichung eines Missbrauchsstoffs oder eines zwanghaften Laufens akkumulieren, eine molekulare Masse von 35-37 kDa. Sie können von der 33-kDa-Isoform von ΔFosB unterschieden werden, die nach einer einzelnen Arzneimittelexposition schnell, aber vorübergehend induziert wird (Abb. 1) (14, 19, 22). Aktuelle Beweise legen nahe, dass die 33-kDa-Isoform die native Form des Proteins ist, das verändert wird, um die stabileren 35- bis 37-kDa-Produkte (19, 21) zu bilden. Die Natur der biochemischen Modifikation, die die instabile 33-kDa-Isoform in die stabilen 35- zu 37-kDa-Isoformen umwandelt, blieb jedoch unklar. Es wurde spekuliert, dass die Phosphorylierung verantwortlich sein könnte (11). Zum Beispiel wird die Induktion von & Dgr; FosB in Mäusen abgeschwächt, denen DARPP-32 fehlt, ein mit Striatum angereichertes Protein (23, 24). Da DARPP-32 die katalytische Aktivität von Proteinphosphatase-1 und Proteinkinase A (25, 26) reguliert, legt die Anforderung an dieses Protein für die normale Akkumulation der stabilen & Dgr; FosB-Isoformen eine mögliche Rolle für die Phosphorylierung bei der Erzeugung dieser stabilen Produkte nahe.
Rolle von ΔFosB in Verhaltensplastizität zu Drogen des Missbrauchs
Der Einblick in die Rolle von ΔFosB in der Drogenabhängigkeit ist hauptsächlich auf transgene Mäuse zurückzuführen, in denen ΔFosB selektiv im Nucleus accumbens und anderen striatalen Regionen adulter Tiere (27, 28) induziert werden kann. Wichtig ist, dass diese Mäuse ΔFosB selektiv in den Dynorphin / Substanz P-enthaltenden Medium-Spiny-Neuronen überexprimieren, wobei angenommen wird, dass die Wirkstoffe das Protein induzieren. Der Verhaltensphänotyp der & Dgr; FosB-überexprimierenden Mäuse, der in vieler Hinsicht Tieren nach chronischer Arzneimittelexposition ähnelt, ist in Tabelle 1 zusammengefasst. Die Mäuse zeigen nach akuter und chronischer Verabreichung (28) verstärkte lokomotorische Reaktionen auf Kokain. Sie zeigen auch eine erhöhte Empfindlichkeit für die Belohnungswirkungen von Kokain und Morphin bei Place-Conditioning-Assays (11, 28) und werden niedrigere Dosen Kokain selbst verabreichen als Wurfgenossen, die ΔFosB nicht überexprimieren. ‡ Im Gegensatz dazu zeigen diese Tiere normal bedingte lokomotorische Zustände Sensibilisierung für Kokain und normales räumliches Lernen im Morris-Wasserlabyrinth (28). TDiese Daten zeigen, dass ΔFosB die Empfindlichkeit eines Tieres gegenüber Kokain und möglicherweise anderen Drogen des Missbrauchs erhöht und einen Mechanismus für eine relativ lange Sensibilisierung gegenüber den Arzneimitteln darstellen kann.
 Striatum
Striatum
| Erhöhte lokomotorische Aktivierung als Reaktion auf akute und wiederholte Kokain-Verabreichung. |
| Verstärkte lohnende Reaktionen auf Kokain und Morphin in Assays zur Ortkonditionierung. |
| Erhöhte Selbstverabreichung von niedrigen Dosen von Kokain. |
| Erhöhte Motivation für Kokain in Progressive-Ratio-Assays. |
| Erhöhte anxiolytische Reaktionen auf Alkohol. |
| Erhöhtes zwanghaftes Laufverhalten. |
Basierend auf Daten in Refs. 28 und 29.† ‡ §¶
Verhaltensplastizität vermittelt durch ΔFosB im Nucleus accumbens-dorsalen Striatum
IDarüber hinaus gibt es vorläufige Hinweise darauf, dass die Wirkungen von ΔFosB weit über eine Regulierung der Medikamentensensitivität per se hin zu komplexeren Verhaltensweisen im Zusammenhang mit dem Suchtprozess hinausgehen können. Mäuse, die & Dgr; FosB exprimieren, arbeiten härter bei der Selbstverabreichung von Kokain in Selbstverabreichungsassays mit progressivem Verhältnis, suEs wird angenommen, dass ΔFosB die Tiere für die Anreiz-Motivationseigenschaften von Kokain sensibilisieren kann und dadurch zu einer Rückfallneigung nach Arzneimittelentzug führen kannl. ‡ ΔFosB-exprimierende Mäuse zeigen auch verstärkte anxiolytische Wirkungen von Alkohol, § ein Phänotyp, der mit einer erhöhten Alkoholaufnahme beim Menschen in Verbindung gebracht wurde. Zusammenfassend deuten diese frühen Ergebnisse darauf hin, dass ΔFosB neben der zunehmenden Sensibilität gegenüber Missbrauchsdrogen auch qualitative Verhaltensänderungen hervorruft, die das Suchtverhalten fördern. Somit kann ΔFosB als ein anhaltender "molekularer Schalter" fungieren, der dabei hilft, entscheidende Aspekte des abhängigen Zustands zu initiieren und dann aufrechtzuerhalten. Eine wichtige Frage, die derzeit untersucht wird, ist, ob die ΔFosB-Akkumulation während der Arzneimittelexposition das Suchtverhalten nach längeren Entzugsperioden fördert, selbst nachdem sich die ΔFosB-Werte normalisiert haben (siehe unten).
Erwachsenen- Mäuse, die ΔFosB selektiv innerhalb des Nucleus accumbens und des dorsalen Striatums überexprimieren, zeigen im Vergleich zu Kontrollkameraden auch eine stärkere Zwangsläufigkeit. Diese Beobachtungen werfen die interessante Möglichkeit auf, dass die ΔFosB-Akkumulation innerhalb dieser Neuronen eine allgemeinere Rolle bei der Bildung und Aufrechterhaltung von Gewohnheitserinnerungen und -zwängen spielt Verhalten, vielleicht durch die Verstärkung der Wirksamkeit von neuronalen Schaltkreisen, in denen diese Neuronen funktionieren.
ΔFosB akkumuliert nach chronischer Kokainexposition in bestimmten Hirnregionen außerhalb des Nucleus accumbens und des dorsalen Striatums. Prominent unter diesen Regionen sind der Amygdala und der mediale präfrontale Kortex (15). Ein Hauptziel der aktuellen Forschung besteht darin, die Beiträge der ΔFosB-Induktion in diesen Regionen zum Suchtphänotyp zu verstehen.
Frühere Arbeiten an fosB-Knockout-Mäusen zeigten, dass diese Tiere keine Sensibilisierung für die lokomotorischen Wirkungen von Kokain entwickeln, was mit den oben erwähnten Befunden der ΔFosB-überexprimierenden Mäuse übereinstimmt (22). Die fosB-Mutanten zeigten jedoch eine erhöhte Empfindlichkeit gegenüber den akuten Wirkungen von Kokain, was mit diesen anderen Befunden nicht vereinbar ist. Die Interpretation der Befunde mit den fosB-Mutanten wird jedoch durch die Tatsache erschwert, dass diesen Tieren nicht nur ΔFosB, sondern auch FosB in voller Länge fehlt. Darüber hinaus fehlen den Mutanten beide Proteine im gesamten Gehirn und in den frühesten Entwicklungsstadien. In der Tat stützen neuere Arbeiten Schlussfolgerungen aus den ΔFosB-überexprimierenden Mäusen: Die induzierbare Überexpression einer verkürzten Mutante von c-Jun, die als dominanter negativer Antagonist von ΔFosB wirkt, zeigt selektiv in Nucleus accumbens und dorsalem Striatum eine verringerte Empfindlichkeit gegenüber den belohnenden Wirkungen von Kokain Diese Ergebnisse unterstreichen die Vorsicht, die bei der Interpretation der Ergebnisse von Mäusen mit konstitutiven Mutationen angewendet werden muss, und veranschaulichen die Bedeutung von Mäusen mit induzierbaren und zelltypspezifischen Mutationen in Studien zur Plastizität im erwachsenen Gehirn.
Zielgene für ΔFosB
Da ΔFosB ein Transkriptionsfaktor ist, verursacht das Protein vermutlich Verhaltensplastizität durch Veränderungen in der Expression anderer Gene. ΔFosB wird durch alternatives Spleißen des fosB-Gens erzeugt und es fehlt ein Teil der C-terminalen Transaktivierungsdomäne, die in FosB voller Länge vorhanden ist. Als Ergebnis wurde ursprünglich vorgeschlagen, dass ΔFosB als Transkriptionsrepressor (29) fungiert. Arbeiten in Zellkultur haben jedoch deutlich gezeigt, dass ΔFosB kann entweder induzieren oder unterdrücken AP-1-vermittelte Transkription abhängig von der jeweils verwendeten AP-1-Stelle (21, 29-31). Full-length FosB übt die gleichen Effekte wie ΔFosB auf bestimmte Promotorfragmente aus, aber andere Effekte auf andere. Weitere Arbeit ist erforderlich, um die Mechanismen zu verstehen, die diesen unterschiedlichen Wirkungen von ΔFosB und FosB zugrunde liegen.
Unsere Gruppe hat zwei Ansätze verwendet, um Zielgene für ΔFosB zu identifizieren. Einer ist der Kandidatengenansatz. Wir betrachteten zunächst α-Amino-3-hydroxy-5-methyl-4-isoxazolpropionsäure (AMPA) -Glutamatrezeptoren als mutmaßliche Ziele, da die glutamaterge Übertragung im Nucleus accumbens eine wichtige Rolle spielt. Bisherige Arbeiten haben gezeigt, dass eine bestimmte AMPA-Glutamatrezeptor-Untereinheit, GluR2, ein echtes Ziel für ΔFosB sein kann (Fig. 2). Die GluR2-Expression, jedoch nicht die Expression anderer AMPA-Rezeptoruntereinheiten, ist im Nucleus accumbens (aber nicht im dorsalen Striatum) bei Überexpression von ΔFosB (28) erhöht, und die Expression einer dominanten negativen Mutante schwächt die Fähigkeit von Kokain ab, das Protein zu induzieren. Zusätzlich enthält der Promotor des GluR2-Gens eine Konsensus-AP-1-Stelle, die ΔFosB bindet (28). Die Überexpression von GluR2 im Nucleus accumbens durch Verwendung eines viral vermittelten Gentransfers erhöht die Empfindlichkeit eines Tieres gegenüber den belohnenden Wirkungen von Kokain und ahmt dadurch einen Teil des Phänotyps nach, der bei den ΔFosB-exprimierenden Mäusen beobachtet wird (28). Die Induktion von GluR2 könnte für die verringerte elektrophysiologische Empfindlichkeit von Nucleus accumbens-Neuronen gegenüber AMPA-Rezeptoragonisten nach chronischer Kokainverabreichung verantwortlich sein (32), da AMPA-Rezeptoren, die GluR2 enthalten, eine verringerte Gesamtleitfähigkeit und eine verringerte Ca2 + -Permeabilität aufweisen. Eine verringerte Reaktion dieser Neuronen auf anregende Eingaben kann dann die Reaktionen auf eine Droge des Missbrauchs verbessern. Die Art und Weise, wie dopaminerge und glutamaterge Signale im Nucleus accumbens das Suchtverhalten regulieren, ist jedoch unbekannt. Dies erfordert ein Verständnis der neuronalen Schaltung, das noch nicht verfügbar ist.
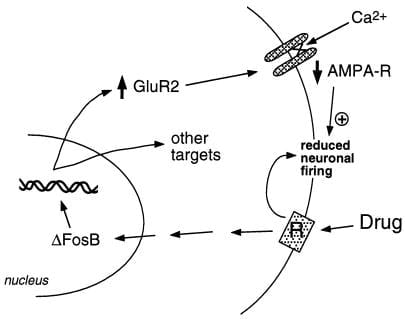
Figure 2
Die AMPA-Glutamatrezeptor-Untereinheit GluR2 ist ein mutmaßliches Ziel für ΔFosB. Dargestellt ist, wie die ΔFosB-vermittelte Induktion von GluR2 die physiologische Reaktionsfähigkeit von Nucleus accumbens-Neuronen verändern und zu sensibilisierten Reaktionen auf Drogenmissbrauch führen kann. Nach diesem Schema erzeugen Drogen des Missbrauchs ihre akute verstärkende Wirkung durch Hemmung der Neuronen des Nucleus accumbens. Bei wiederholter Exposition induzieren die Medikamente ΔFosB, das zahlreiche Zielgene, einschließlich GluR2, reguliert. Dies erhöht den Anteil der AMPA-Rezeptoren (AMPA-R) an Nucleus accumbens-Neuronen, die die GluR2-Untereinheit enthalten, was einen verringerten Gesamt-AMPA-Strom und einen verringerten Ca2 + -Strom verursacht. Diese verringerte Erregbarkeit könnte die Neuronen empfindlicher für die akuten Hemmwirkungen der Arzneimittel und damit für die verstärkenden Wirkungen der Arzneimittel machen.
Ein anderes mutmaßliches Ziel für & Dgr; FosB ist das Gen, das für Dynorphin kodiert. Wie zuvor erwähnt, wird Dynorphin in der Untergruppe von Nucleus accumbens medium stacheligen Neuronen exprimiert, die eine Induktion von ΔFosB zeigen. Dynorphin scheint in einer interzellulären Rückkopplungsschleife zu funktionieren: seine Freisetzung inhibiert die dopaminergen Neuronen, die die mittleren stacheligen Neuronen innervieren, über κ Opioidrezeptoren, die an dopaminergen Nervenenden im Nucleus accumbens vorhanden sind, und auch an Zellkörpern und Dendriten im ventralen Tegmentum (Abb. 3) (33-35). Diese Idee stimmt mit der Fähigkeit eines & kgr; -Rezeptor-Agonisten überein, nach Verabreichung in eine dieser beiden Gehirnregionen die Wiederbelebung des Arzneimittels zu verringernd (35).
RE-Arbeiten haben gezeigt, dass ΔFosB die Expression von Dynorphin verringert, was zur Verbesserung der Belohnungsmechanismen beitragen könnte, die bei der ΔFosB-Induktion beobachtet werden. Interessanterweise bewirkt ein weiterer arzneimittelregulierter Transkriptionsfaktor, CREB (cAMP-Response-Element-bindendes Protein) (2, 3), den gegenteiligen Effekt: Er induziert eine Dynorphin-Expression im Nucleus accumbens und reduziert die Belohnungseigenschaften von Kokain und Morphin (4). **
BDa die medikamenteninduzierte Aktivierung von CREB schnell nach der Verabreichung des Medikaments abklingt, könnte eine solche reziproke Regulation von Dynorphin durch CREB und ΔFosB die reziproken Verhaltensänderungen während früher und später Phasen des Entzugs erklären, wobei negative emotionale Symptome und eine reduzierte Arzneimittelsensitivität in frühen Phasen überwiegen des Entzugs und der Sensibilisierung für die Belohnungs- und Anreizmotivationseffekte von Drogen, die zu späteren Zeitpunkten überwiegen.
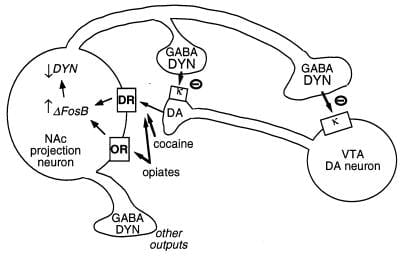
Figure 3
Dynorphin ist ein mutmaßliches Ziel für ΔFosB. Dargestellt ist ein Dopamin (DA) -Neuron des ventralen tegmentalen Bereichs (VTA), das eine Klasse von Nukleus Accumbens (NAc) GABAergenes Projektionsneuron innerviert, das Dynorphin (DYN) exprimiert. Dynorphin dient einem Rückkopplungsmechanismus in dieser Schaltung: Dynorphin, das von den Enden der NAc-Neuronen freigesetzt wird, wirkt auf κ-Opioidrezeptoren, die sich auf Nervenenden und Zellkörpern der DA-Neuronen befinden, um deren Funktion zu hemmen. ΔFosBkann durch Hemmung der Dynorphin-Expression diese Rückkopplungsschleife herunterregulieren und die Belohnungseigenschaften von Missbrauchsdrogen verbessern. Nicht gezeigt ist die reziproke Wirkung von CREB auf dieses System: CREB verstärkt die Dynorphin-Expression und vermindert dadurch die Belohnungseigenschaften von Missbrauchsdrogen (4). GABA, γ-Aminobuttersäure; DR, Dopaminrezeptor; ODER, Opioidrezeptor.
Der zweite Ansatz zur Identifizierung von Zielgenen für ΔFosB umfasst die DNA-Microarray-Analyse. Die induzierbare Überexpression von ΔFosB erhöht oder verringert die Expression zahlreicher Gene im Nucleus accumbens (36). Obwohl jetzt beträchtliche Arbeit erforderlich ist, um jedes dieser Gene als physiologische Ziele von ΔFosB zu validieren und ihren Beitrag zum Suchtphänotyp zu verstehen, scheint ein wichtiges Ziel Cdk5 (Cyclin-abhängige Kinase-5) zu sein. Daher wurde Cdk5 zunächst unter Verwendung von Microarrays als ΔFosB-reguliert identifiziert und später nach chronischer Kokainverabreichung im Nucleus accumbens und im dorsalen Striatum induziert (37). ΔFosB aktiviert das cdk5-Gen über eine AP-1-Stelle, die im Promotor des Gens vorhanden ist (36). Zusammen unterstützen diese Daten ein Schema, bei dem Kokain die Cdk5-Expression in diesen Hirnregionen über ΔFosB induziert. Die Induktion von Cdk5 scheint die dopaminerge Signalübertragung zumindest teilweise über eine erhöhte Phosphorylierung von DARPP-32 (37) zu verändern, das bei seiner Phosphorylierung durch Cdk1 von einem Inhibitor der Proteinphosphatase-5 in einen Inhibitor der Proteinkinase A umgewandelt wird (26).
Die Rolle von ΔFosB bei der Vermittlung von "permanenter" Plastizität an Missbrauchsdrogen
Obwohl das ΔFosB-Signal relativ langlebig ist, ist es nicht permanent. ΔFosB degradiert allmählich und ist nach 1-2 Monaten des Drogenentzugs im Gehirn nicht mehr nachweisbar, obwohl bestimmte Verhaltensanomalien über viel längere Zeit bestehen bleiben. Daher scheint ΔFosB per se nicht in der Lage zu sein, diese semipermanenten Verhaltensauffälligkeiten zu vermitteln. Die Schwierigkeit, die molekularen Anpassungen zu finden, die den extrem stabilen Verhaltensänderungen im Zusammenhang mit der Sucht zugrunde liegen, ist analog zu den Herausforderungen, die sich im Bereich Lernen und Gedächtnis stellen. Obwohl es elegante zelluläre und molekulare Modelle des Lernens und des Gedächtnisses gibt, war es bisher nicht möglich, molekulare und zelluläre Anpassungen zu identifizieren, die ausreichend langlebig sind, um hochstabile Verhaltensspeicher zu erklären. In der Tat ist ΔFosB die langlebigste Anpassung, von der bekannt ist, dass sie im erwachsenen Gehirn auftritt, nicht nur als Reaktion auf Drogenmissbrauch, sondern auch auf jede andere Störung (die keine Läsionen beinhaltet). Zwei Vorschläge haben sich sowohl im Bereich Sucht als auch im Bereich Lernen und Gedächtnis entwickelt, um diese Diskrepanz zu erklären.
Eine Möglichkeit ist, dass transiente Veränderungen der Genexpression, wie sie über ΔFosB oder andere Transkriptionsfaktoren (zB CREB) vermittelt werden, kann länger anhaltende Veränderungen in der neuronalen Morphologie und der synaptischen Struktur vermitteln. Zum Beispiel, eine Zunahme der Dichte von dendritischen Stacheln (insbesondere eine Zunahme von zweiköpfigen Stacheln) begleitet die erhöhte Wirksamkeit von glutamatergen Synapsen an Hippocampus-Pyramidenneuronen während der Langzeitpotenzierung (38-40) und parallel zu der erhöhten Verhaltensempfindlichkeit gegenüber Kokain, die auf der Ebene der mittleren stacheligen Neuronen des Nucleus accumbens (41) vermittelt wird. Es ist nicht bekannt, ob solche strukturellen Veränderungen ausreichend langlebig sind, um hochstabile Verhaltensänderungen zu berücksichtigen, obwohl letztere mindestens für den 1-Monat des Drogenentzugs bestehen bleiben. Neuere Befunde lassen vermuten, dass ΔFosB und seine Induktion von Cdk5 ein Vermittler von arzneimittelinduzierten Veränderungen der synaptischen Struktur im Nucleus accumbens ist (Abb. 4). ‡ ‡ Daher verhindert die Infusion eines Cdk5-Inhibitors in den Nucleus accumbens Fähigkeit einer wiederholten Kokainexposition, die dendritische Wirbelsäulendichte in dieser Region zu erhöhen. Dies stimmt mit der Ansicht überein, dass Cdk5, das im Gehirn angereichert ist, die neurale Struktur und das Wachstum reguliert (siehe Ref. 36 und 37). Es ist möglich, obwohl keineswegs bewiesen, dass solche Veränderungen in der neuronalen Morphologie das ΔFosB-Signal selbst überdauern können.
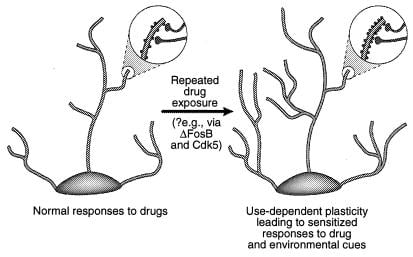
Figure 4
Regulierung der dendritischen Struktur durch Drogenmissbrauch. Dargestellt ist die Ausdehnung des dendritischen Baums eines Neurons nach chronischer Exposition gegenüber einer Droge des Missbrauchs, wie dies bei Kokain im Nucleus accumbens und im präfrontalen Kortex beobachtet wurde (41). Die Vergrößerungsbereiche zeigen eine Zunahme der dendritischen Stacheln, von der postuliert wird, dass sie in Verbindung mit aktivierten Nervenenden auftritt. Diese Zunahme der dendritischen Wirbelsäulendichte kann über ΔFosB und die daraus resultierende Induktion von Cdk5 vermittelt werden (siehe Text). Solche Veränderungen in der dendritischen Struktur, die denen ähneln, die in einigen Lernmodellen beobachtet wurden (z. B. Langzeitpotenzierung), könnten langlebige sensibilisierte Reaktionen auf Drogenmissbrauch oder Umwelteinflüsse vermitteln. [Wiedergabe mit freundlicher Genehmigung aus Lit. 3 (Copyright 2001, Macmillian Magazines Ltd.)].
Eine andere Möglichkeit ist die transiente Induktion eines Transkriptionsfaktors (zB ΔFosB, CREB). führt zu dauerhaften Veränderungen in der Genexpression durch die Modifikation von Chromatin. Es wird angenommen, dass diese und viele andere Transkriptionsfaktoren die Transkription eines Zielgens aktivieren oder unterdrücken, indem sie die Acetylierung bzw. Deacetylierung von Histonen in der Nähe des Gens (42) fördern. Obwohl eine solche Acetylierung und Deacetylierung von Histonen anscheinend sehr schnell stattfinden kann, ist es möglich, dass & Dgr; FosB oder CREB längeranhaltende Anpassungen in der enzymatischen Maschinerie, die die Histonacetylierung steuert, erzeugen können. ΔFosB oder CREB können auch länger anhaltende Veränderungen der Genexpression fördern, indem sie andere Modifikationen des Chromatins (z. B. DNA- oder Histonmethylierung) regulieren, die an den permanenten Veränderungen der Gentranskription während der Entwicklung beteiligt sind (siehe Lit. 42 und 43). . Obwohl diese Möglichkeiten spekulativ bleiben, könnten sie einen Mechanismus bereitstellen, durch den vorübergehende Anpassungen an ein Missbrauchsdrogen (oder eine andere Störung) zu im Wesentlichen lebenslangen Verhaltenskonsequenzen führen.
Referenzen
- ↵
- Nestler EJ,
- Hoffnung BT,
- Widnell KL
(1993) Neuron 11: 995-1006.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Berke JD,
- Hyman SE
(2000) Neuron 25: 515-532.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Nestler EJ
(2001) Nat Rev Neurosci 2: 119-128.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Carlezon WA Jr,
- Thome J,
- Olson VG,
- Lane-Ladd SB,
- Brodkin ES,
- Hiroi N,
- Duman RS,
- Neve RL,
- Nestler EJ
(1998) Wissenschaft 282: 2272-2275.
Zusammenfassung / KOSTENLOSER Volltext
-
- O'Donovan KJ,
- Tourtellotte WG,
- Millbrandt J,
- Baraban JM
(1999) Trends Neurosci 22: 167-173.
- ↵
- Mackler SA,
- Korutla L,
- Cha XY,
- Koebbe MJ,
- Fournier KM,
- Bowers MS,
- Kalivas PW
(2000) J Neurosc. 20: 6210-6217.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Morgan JI,
- Curran T
(1995) Trends Neurosci 18: 66-67.
- ↵
- Junge ST,
- Porrino LJ,
- Iadarola MJ
(1991) Proc Natl Acad Sci Deutschland 88: 1291-1295.
Zusammenfassung / KOSTENLOSER Volltext
-
- Graybiel AM,
- Moratalla R,
- Robertson HA
(1990) Proc Natl Acad Sci Deutschland 87: 6912-6916.
Zusammenfassung / KOSTENLOSER Volltext
-
- Hoffnung B,
- Kosofsky B,
- Hyman SE,
- Nestler EJ
(1992) Proc Natl Acad Sci Deutschland 89: 5764-5768.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Kelz MB,
- Nestler EJ
(2000) Curr Opin Neurol 13: 715-720.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Koob GF,
- Sanna PP,
- Blüte FE
(1998) Neuron 21: 467-476.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Kluge RA
(1998) Drogenabhängigkeit 51: 13-22.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Hoffnung BT,
- Nye HE,
- Kelz MB,
- Selbst DW,
- Iadarola MJ,
- Nakabeppu Y,
- Duman RS,
- Nestler EJ
(1994) Neuron 13: 1235-1244.
- ↵
- Nye H,
- Hoffnung BT,
- Kelm M,
- Iadarola M,
- Nestler EJ
(1995) J Pharmacol Exp Ther 275: 1671-1680.
Zusammenfassung / KOSTENLOSER Volltext
-
- Nye HE,
- Nestler EJ
(1996) Mol Pharmacol 49: 636-645.
- ↵
- Moratalla R,
- Elibol B,
- Vallejo M,
- Graybiel AM
(1996) Neuron 17: 147-156.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Pich EM,
- Pagliusi SR,
- Tessari M,
- Talabot-Ayer D,
- Hooft van Huijsduijnen R,
- Chiamulera C
(1997) Wissenschaft 275: 83-86.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Chen JS,
- Nye HE,
- Kelz MB,
- Hiroi N,
- Nakabeppu Y,
- Hoffnung BT,
- Nestler EJ
(1995) Mol Pharmacol 48: 880-889.
- ↵
- Hiroi N,
- Brauner J,
- Ye H,
- Saudou F,
- Vaidya VA,
- Duman RS,
- Greenberg ME,
- Nestler EJ
(1998) J Neurosc. 18: 6952-6962.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Chen J,
- Kelz MB,
- Hoffnung BT,
- Nakabeppu Y,
- Nestler EJ
(1997) J Neurosc. 17: 4933-4941.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Hiroi N,
- Brauner J,
- Haile C,
- Ye H,
- Greenberg ME,
- Nestler EJ
(1997) Proc Natl Acad Sci Deutschland 94: 10397-10402.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Fienberg AA,
- Hiroi N,
- Mermelstein P,
- Lied WJ,
- Snyder GL,
- Nishi A,
- Cheramy A,
- O'Callaghan JP,
- Miller D,
- Cole DG,
- et al.
(1998) Wissenschaft 281: 838-842.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Hiroi N,
- Feinberg A,
- Haile C,
- Greengard P,
- Nestler EJ
(1999) Eur J Neurosc. 11: 1114-1118.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Greengard P,
- Allen PB,
- Nairn AC
(1999) Neuron 23: 435-447.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Bibb JA,
- Snyder GL,
- Nishi A,
- Yan Z,
- Meijer L,
- Fienberg AA,
- Tsai LH,
- Kwon YT,
- Girault JA,
- Czernik AJ,
- et al.
(1999) Natur (London) 402: 669-671.
- ↵
- Chen JS,
- Kelz MB,
- Zeng GQ,
- Sakai N,
- Steffen C,
- Shockett PE,
- Picciotto M,
- Duman RS,
- Nestler EJ
(1998) Mol Pharmacol 54: 495-503.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Kelz MB,
- Chen JS,
- Carlezón WA,
- Whisler K,
- Gilden L,
- Beckmann AM,
- Steffen C,
- Zhang YJ,
- Marotti L,
- Selbst SW,
- et al.
(1999) Natur (London) 401: 272-276.
- ↵
- Dobrazanski P,
- Noguchi T,
- Kovary K,
- Rizzo CA,
- Lazo PS,
- Bravo R
(1991) Mol Zelle Biol 11: 5470-5478.
Zusammenfassung / KOSTENLOSER Volltext
-
- Nakabeppu Y,
- Nathan D
(1991) Zelle 64: 751-759.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Yen J,
- Weisheit RM,
- Tratner ich,
- Verma IM
(1991) Proc Natl Acad Sci Deutschland 88: 5077-5081.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Weiß FJ,
- Hu XT,
- Zhang XF,
- Wolf ME
(1995) J Pharmacol Exp Ther 273: 445-454.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Hyman SE
(1996) Neuron 16: 901-904.
-
- Kreek MJ
(1997) Pharmacol Biochem Verhalten 57: 551-569.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Shippenberg TS,
- Rea W
(1997) Pharmacol Biochem Verhalten 57: 449-455.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Chen JS,
- Zhang YJ,
- Kelz MB,
- Steffen C,
- Ang ES,
- Zeng L,
- Nestler EJ
(2000) J Neurosc. 20: 8965-8971.
Zusammenfassung / KOSTENLOSER Volltext
- ↵
- Bibb JA,
- Chen JS,
- Taylor JR,
- Svenningsson P,
- Nishi A,
- Snyder GL,
- Yan Z,
- Sagawa ZK,
- Nairn Wechselstrom,
- Nestler EJ,
- et al.
(2001) Natur (London) 410: 376-380.
- ↵
- Luscher C,
- Nicoll RA,
- Malenka RC,
- Müller D
(2000) Nat Neurosci 3: 545-550.
CrossRefMedlineWeb der Wissenschaft
-
- Malinow R,
- Mainen ZF,
- Hayashi Y
(2000) Curr Opin Neurobiol 10: 352-357.
CrossRefMedlineWeb der Wissenschaft
- ↵
- Scannevin RH,
- Huganir RL
(2000) Nat Rev Neurosci 1: 133-141.
CrossRefMedlineWeb der Wissenschaft
Robinson, TE & Kolb, B. (1999) (1997) EUR. J. Neurosci.11, 1598-1604.
- ↵
- Carey M,
- Smale ST
(2000) Transkriptionsregulation in Eukaryoten (Cold Spring Harbor Lab. Press, Plainview, NY).
- ↵
- Spencer VA,
- Davie JR
(1999) Gen 240: 1-12.
CrossRefMedlineWeb der Wissenschaft
 Facebook
Facebook Twitter
Twitter- Google+
 CiteULike
CiteULike köstlich
köstlich Digg
Digg Mendeley
Mendeley
In der HighWire Press gehostete Artikel zitieren diesen Artikel
- Natural and Drug Rewards Gesetz über gemeinsame Mechanismen der neuronalen Plastizität mit {FosB} als Schlüsselvermittler J. Neurosci. 2013 33 (8) 3434-3442
- Drogen, Verbrechen und die Epigenetik der Hedonischen Allostasis Zeitschrift für zeitgenössische Strafjustiz 2012 28 (3) 314-328
- Abstract
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Abstract
- Volltext (HTML)
- Volltext (PDF)
- Morphin aktiviert den E-26-ähnlichen Transkriptionsfaktor-1 / Serum-Response-Faktor-Weg über extrazelluläre signalregulierte Kinasen 1 / 2 in F11-Zellen, die von Neuronen der Dorsalwurzel-Ganglien abgeleitet sind J. Pharmacol. Exp. Ther. 2012 342 (1) 41-52
- Molekularer Mechanismus für ein Gateway-Medikament: Epigenetische Veränderungen, initiiert durch Nikotin-Prime-Genexpression durch Kokain Sci Translation 2011 3 (107) 107ra109
- Verbesserte Sucrose- und Kokain-Selbstverwaltung und Cue-induzierte Arzneimittelsuche nach Verlust von VGLUT2 in Mittelhirn-Dopamin-Neuronen in Mäusen J. Neurosci. 2011 31 (35) 12593-12603
- Chronische intermittierende Hypoxie erhöht den Blutdruck und die Expression von FosB / FosB in zentralen autonomen Regionen Bin ich J. Physiol. Regul. Integr. Komp. Physiol. 2011 301 (1) R131-R139
- Das Fehlen des GPR37 / PAEL-Rezeptors beeinträchtigt die striatale Akt- und ERK2-Phosphorylierung, die δFosB-Expression und die konditionierte Ortspräferenz gegenüber Amphetamin und Kokain FASEB J. 201125 (6) 2071-2081
- Die Beziehung zwischen der Dauer der anfänglichen Alkoholexposition und der Beständigkeit der molekularen Toleranz ist deutlich nichtlinear J. Neurosci. 2011 31 (7) 2436-2446
- In-vivo-Biolumineszenz-Bildgebung zeigt eine Redox-regulierte Aktivator-Protein-1-Aktivierung im paraventrikulären Nucleus von Mäusen mit renovaskulärer Hypertonie Bluthochdruck 2011 57 (2) 289-297
- Striatale Überexpression von {FosB} reproduziert chronische Levodopa-induzierte unwillkürliche Bewegungen J. Neurosci. 2010 30 (21) 7335-7343
- Epigenetische Mediation von Umwelteinflüssen bei schweren psychotischen Störungen Schizophr Stier 2009 35 (6) 1045-1056
- DNA-basierte MRT-Sonden für den spezifischen Nachweis einer chronischen Exposition gegenüber Amphetamin in lebenden Gehirnen J. Neurosci. 2009 29 (34) 10663-10670
- Veränderte dendritische Wirbelsäulenplastizität in Kokain-Zurückgezogenen Ratten J. Neurosci. 2009 29 (9) 2876-2884
- Überexpressions-Screen in Drosophila identifiziert neuronale Rollen von GSK-3 {beta} / shaggy als Regulator der AP-1-abhängigen Entwicklungsplastizität Genetik 2008 180 (4) 2057-2071
- Transkription MRT: Eine neue Sicht auf das lebende Gehirn Neurowissenschaftler 2008 14 (5) 503-520
- {F} B-Induktion im orbitofrontalen Kortex vermittelt Toleranz gegenüber Kokain-induzierter kognitiver Dysfunktion J. Neurosci. 2007 27 (39) 10497-10507
- Endgültige Anfälligkeit für die Wiederherstellung des Methamphetamin-Suchverhaltens in aus Gliazelllinien stammenden neurotrophen Faktor-mutierten Mäusen FASEB J. 200721 (9) 1994-2004
- {FosB} im Nucleus Accumbens reguliert das nahrungsmittelverstärkte instrumentelle Verhalten und die Motivation J. Neurosci. 2006 26 (36) 9196-9204
- Regulation der {F} B-Stabilität durch Phosphorylierung. J. Neurosci. 2006 26 (19) 5131-5142
- Expression von Mutanten-NMDA-Rezeptoren in Dopamin-D1-Rezeptor-enthaltenden Zellen verhindert Kokain-Sensibilisierung und verringert Kokain-Präferenz J. Neurosci. 2005 25 (28) 6651-6657
- D1-Dopaminrezeptoren modulieren die {F} B-Induktion in Rattenstriatum nach intermittierender Morphin-Verabreichung J. Pharmacol. Exp. Ther. 2005 314 (1) 148-154
- Neurobiologie von Mäusen, ausgewählt für hohe freiwillige Radlaufaktivität Integr. Komp. Biol. 2005 45 (3) 438-455
- Auswirkungen von Wasserentzug und Rehydratisierung auf die c-Fos- und FosB-Färbung im supraoptischen Nucleus der Ratte und der Lamina terminalis-Region Bin ich J. Physiol. Regul. Integr. Komp. Physiol. 2005 288 (1) R311-R321
- Transkriptionsinduktion von FosB / {F} B-Gen durch mechanische Belastung in Osteoblasten J Biol Chem 2004 279 (48) 49795-49803
- Induktion von {F} B in Reward-Related Brain Structures nach chronischem Stress J. Neurosci. 2004 24 (47) 10594-10602
- Die Sim1-Gendosis moduliert die Reaktion der homöostatischen Ernährung auf erhöhte Nahrungsfettwerte bei Mäusen Bin ich J. Physiol. Endocrinol. Metab. 2004 287 (1) E105-E113
- DNA-Microarray-Analyse der Genexpression in menschlichen Sehnervenkopf-Astrozyten als Reaktion auf hydrostatischen Druck Physiol. Genomik 2004 17 (2) 157-169
- Superoxid ist an der Aktivierung des zentralen Nervensystems und an der Sympathoexpression von Myokardinfarkt-induzierter Herzinsuffizienz beteiligt Circ. Res. 2004 94 (3) 402-409
- Adenosin-A2A-Rezeptoren bei der Neuroadaptation an wiederholte dopaminerge Stimulation: Implikationen für die Behandlung von Dyskinesien bei Parkinson Neurologie 2003 61 (90116) S74-81
- Cytoplasmatische versus Kernlokalisierung von Fos-verwandten Proteinen im Frosch, Rana esculenta, Hoden: In vivo und direkte In-vitro-Wirkung eines Gonadotropin-freisetzenden Hormon-Agonisten Biol. Reprod. 2003 68 (3) 954-960
- Periadoleszente Mäuse zeigen eine verstärkte Delta-FosB-Hochregulation in Reaktion auf Kokain und Amphetamin J. Neurosci. 2002 22 (21) 9155-9159
- Delta FosB reguliert den Radlauf J. Neurosci. 2002 22 (18) 8133-8138
- Die CREB-Aktivität in der Nucleus accumbens-Shell steuert das Gating von Verhaltensantworten auf emotionale Stimuli Proc. Natl. Acad. Sci. USA 2002 99 (17) 11435-11440
- Psychogenomik: Möglichkeiten zum Verständnis von Sucht J. Neurosci. 2001 21 (21) 8324-8327